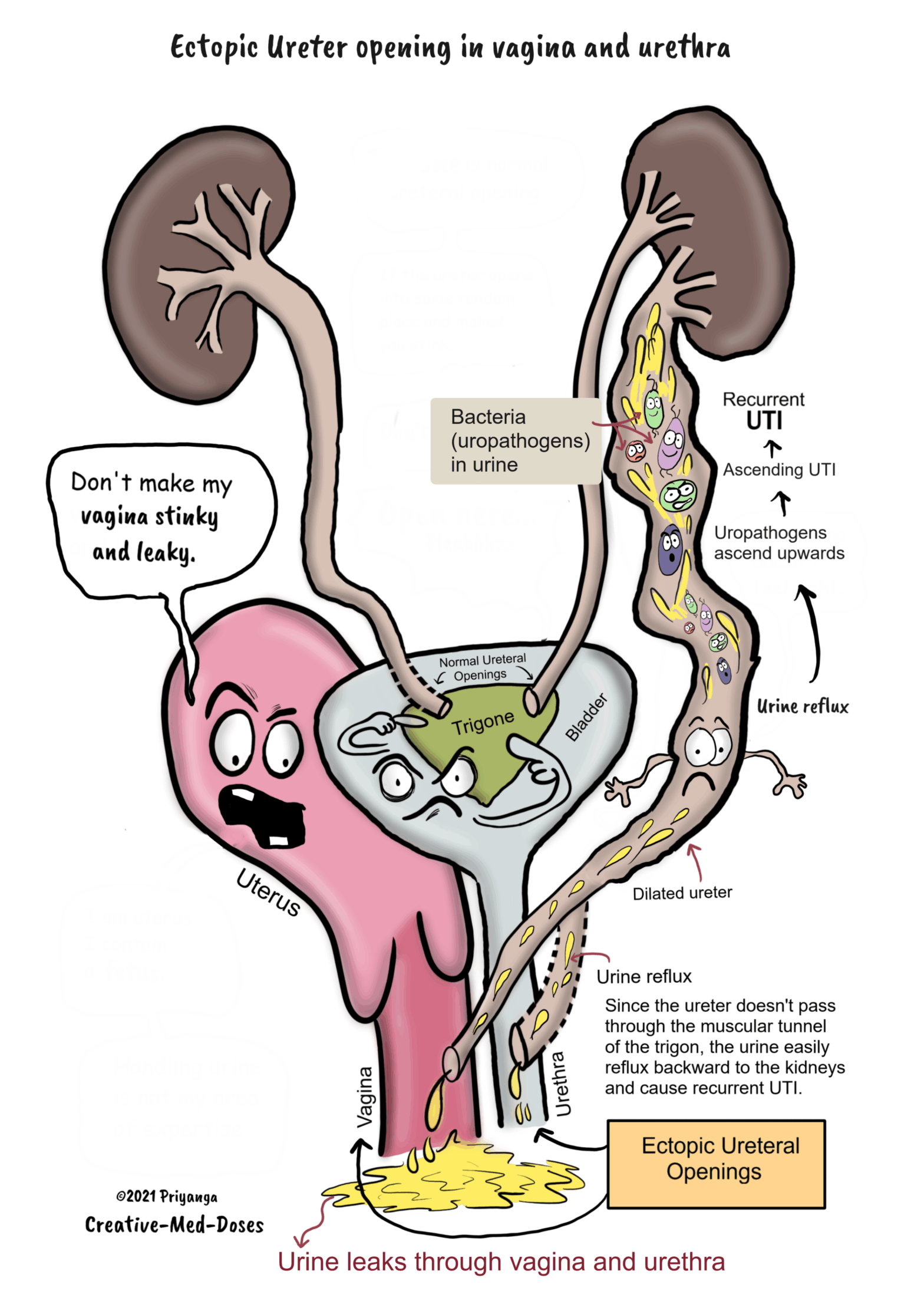
Ectopic ureter
The ectopic ureter is any ureter, single or duplex, which does not open in the trigonal region of the bladder. Nearly 80% of ectopic ureters are associated with complete duplication of the collecting system.
Most common sites of ectopic opening –
In males, this site is usually the urethra, and in females, the ectopic ureter opening is often at the urethra or vagina.
Problems associated with ectopic ureter –
• Often asymptomatic in males
• Vesicoureteral reflux
• Prenatal hydronephrosis
• Recurrent urinary tract infections
• Incontinence – In females, the ectopic ureteric opening may be at the bladder neck, urethra, vagina, or vestibule. The ectopic ureter drains either distal to the urethral sphincter or into the reproductive tract resulting in continuous incontinence.
The toilet-trained girl with normal voiding habits presents with continuous dampness of the genital region. And, gives no history of diurnal variation or increased nighttime bedwetting is a classical presentation of the ectopic ureter with incontinence.
Post Type Archives: Topics
Phosphate binders in chronic kidney disease

Phosphate binders in chronic kidney disease
Phosphate binders (PB) in conjunction with dietary phosphate restriction reduce serum phosphate levels in patients with chronic kidney disease.
Phosphate-restricted diet
The restriction of processed foods, fast foods, dairy products, and cola beverages reduces dietary phosphate intake.
Phosphate binder: Types
PB is used in conjunction with dietary phosphate restriction to achieve the goal of low serum phosphate levels.
Mechanism of action
As the name suggests, the phosphate binders bind to the phosphate in the gastrointestinal tract. The phosphate present in food reaches the intestine to get absorbed into the bloodstream, but the phosphate binders bind to the phosphate, forming an insoluble complex. This insoluble complex cannot be absorbed into the bloodstream and is excreted through the feces. This way, phosphate binders significantly reduce the intestinal absorption of phosphate, resulting in lower blood potassium levels. Phosphate binders are taken orally with a meal or snack so that they can reduce the absorption of phosphate present in the food ingested.
Types of Phosphate binders
• Aluminium containing phosphate binders
• Calcium-containing phosphate binders
• Non-calcium-based phosphate binders
Chronic Kidney Disease-sign and symptoms part 1
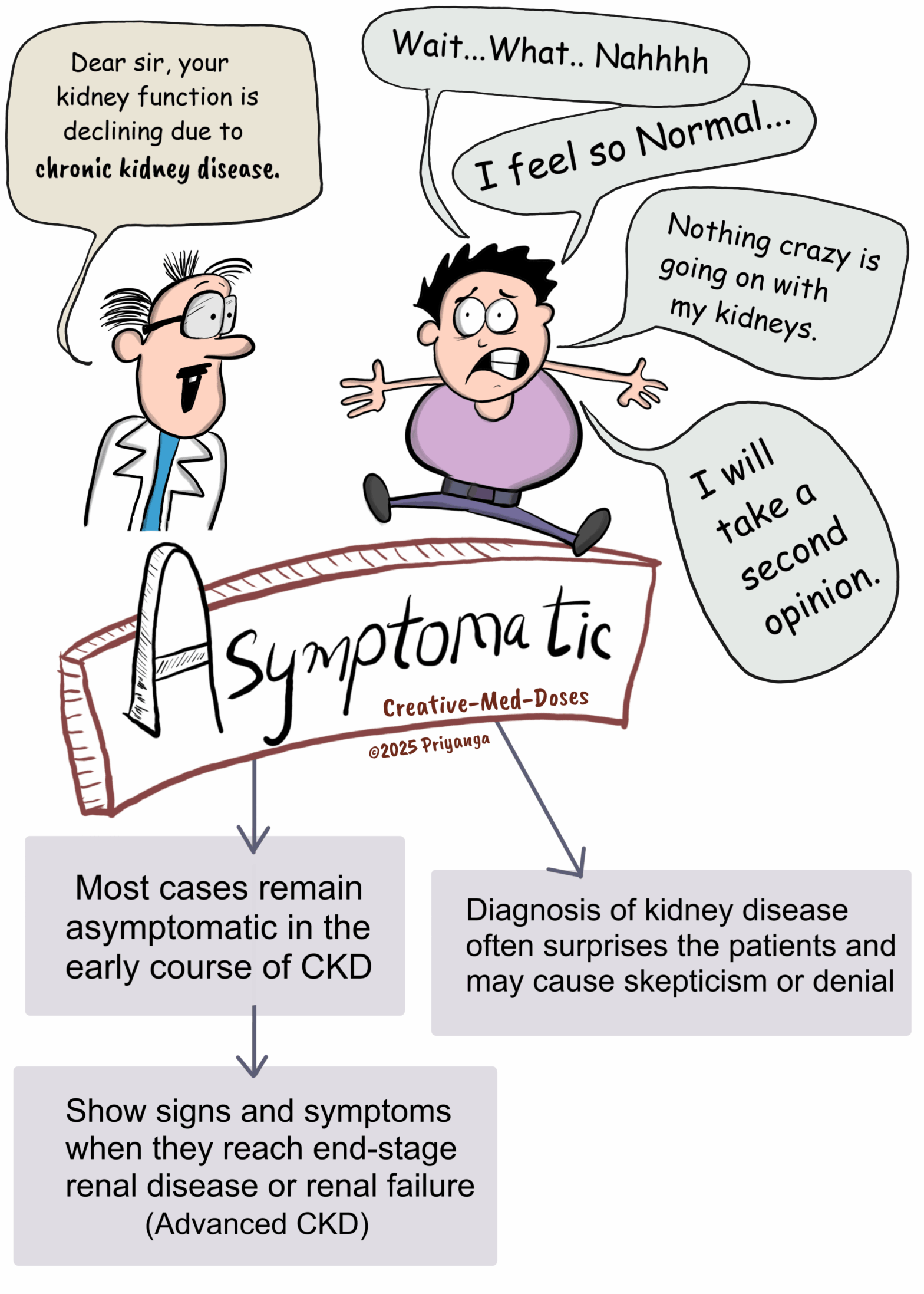
Asymptomatic
– Most cases remain asymptomatic in the early course of CKD
– Show signs and symptoms when they reach end-stage renal disease or renal failure
– Diagnosis of kidney disease often surprises the patients and may cause skepticism or denial
Arteriovenous fistula in renal biopsy

Arteriovenous fistula in renal biopsy is a rare complication of percutaneous renal biopsy.
If the nearby blood vessels (arteries/veins) are injured during the renal biopsy procedures, an anatomical connection between the artery and vein can be formed, known as an arteriovenous fistula.
These connections are usually small, often remain asymptomatic, and resolve spontaneously with time.
Rarely, large A-V fistulas are formed and cause the following symptoms
• Hematuria (blood in urine)
• Drop in the blood pressure
• High-output cardiac failure
Management
Small fistula – observation and follow-up
Larger symptomatic fistula
• Embolization to close A-V fistula
• Arterial ligation
• Nephrectomy in rare cases
Contrast-Induced Nephropathy
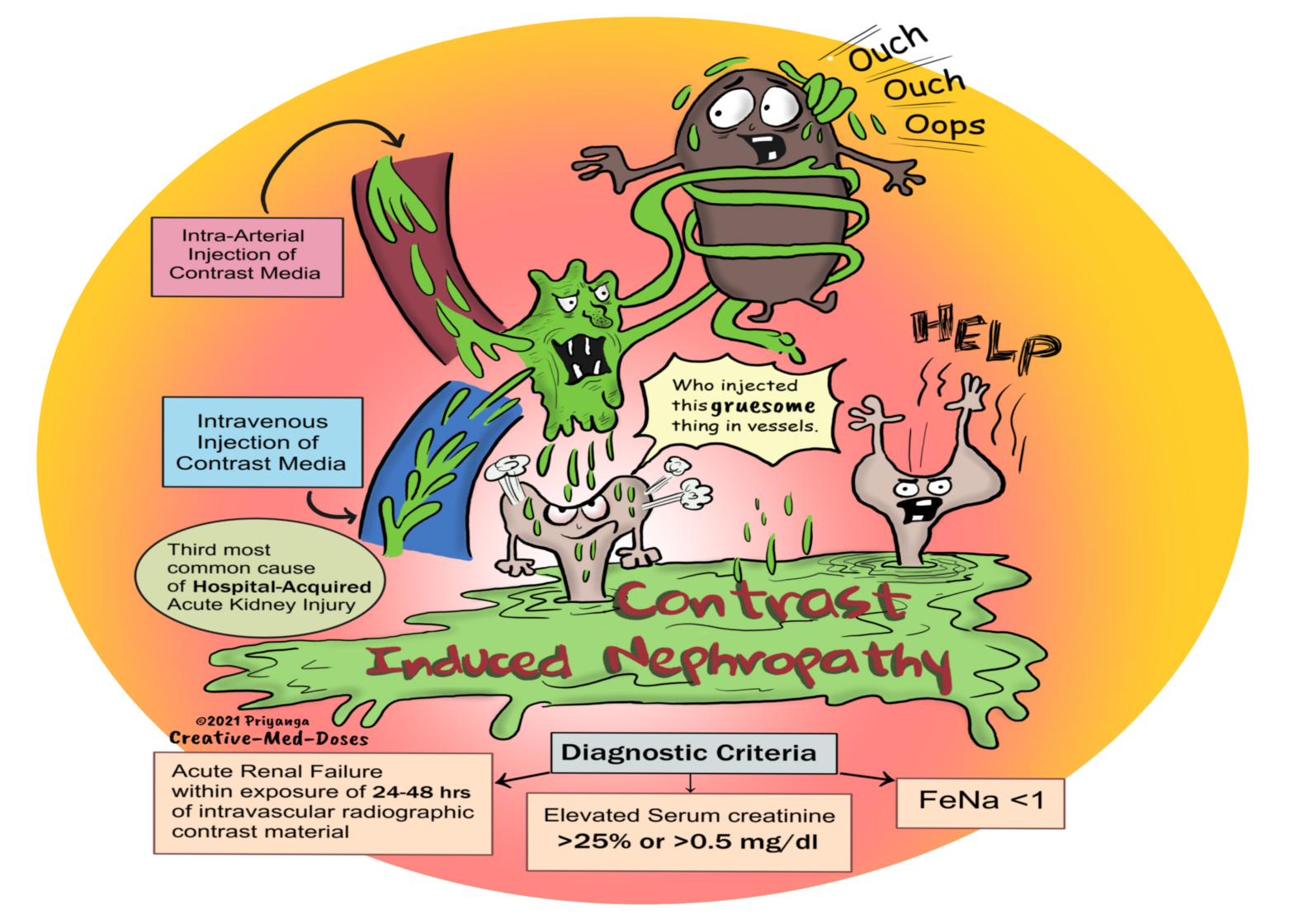
Contrast-induced nephropathy is an acute renal failure occurring within 24-48 hours of exposure to intravascular radiographic contrast material not related to other causes.
It is the third most common cause of hospital-acquired acute renal injury. The first and second cause is surgery and hypotension, respectively.
Diagnostic criteria-
• Rise in serum creatinine (Scr) levels of more than 25% or ≥0.5 mg/dl (44 μmol/l) from baseline within 48 h of contrast media exposure with the exclusion of other causes of acute kidney injury
• Signs of acute tubular necrosis
• Fractional excretion of sodium (FeNA) less than 1
Risk factors
• Chronic Kidney Disease or preexisting impairment of renal function is the most common risk factor.
• Diabetic nephropathy
• Proteinuria- the excess protein in tubular lumen accumulates with contrast media and causes tubular obstruction. Hypoalbuminemia increases free drug concentration in the blood that increases the risk of nephrotoxicity. (clue: most drugs are bound to albumin).
• Multiple Myeloma -It has high amounts of protein in the tubular lumen. The concomitant use of contrast material causes the accumulation of precipitated contrast media and myeloma protein in the tubular lumen. This accumulated protein, contrast media, and desquamated apoptotic tubular cells together cause tubular obstruction and acute tubular injury (obstructive nephropathy).
• Reduced intravascular volume (congestive heart failure, liver cirrhosis, or abnormal fluid losses) causes prolonged hypotension. It contributes to a prerenal reduction in renal perfusion.
• High Osmolality Contrast Media
• Amount of contrast Media
Sodium-glucose cotransporter-2 inhibitors (SGLT2i)
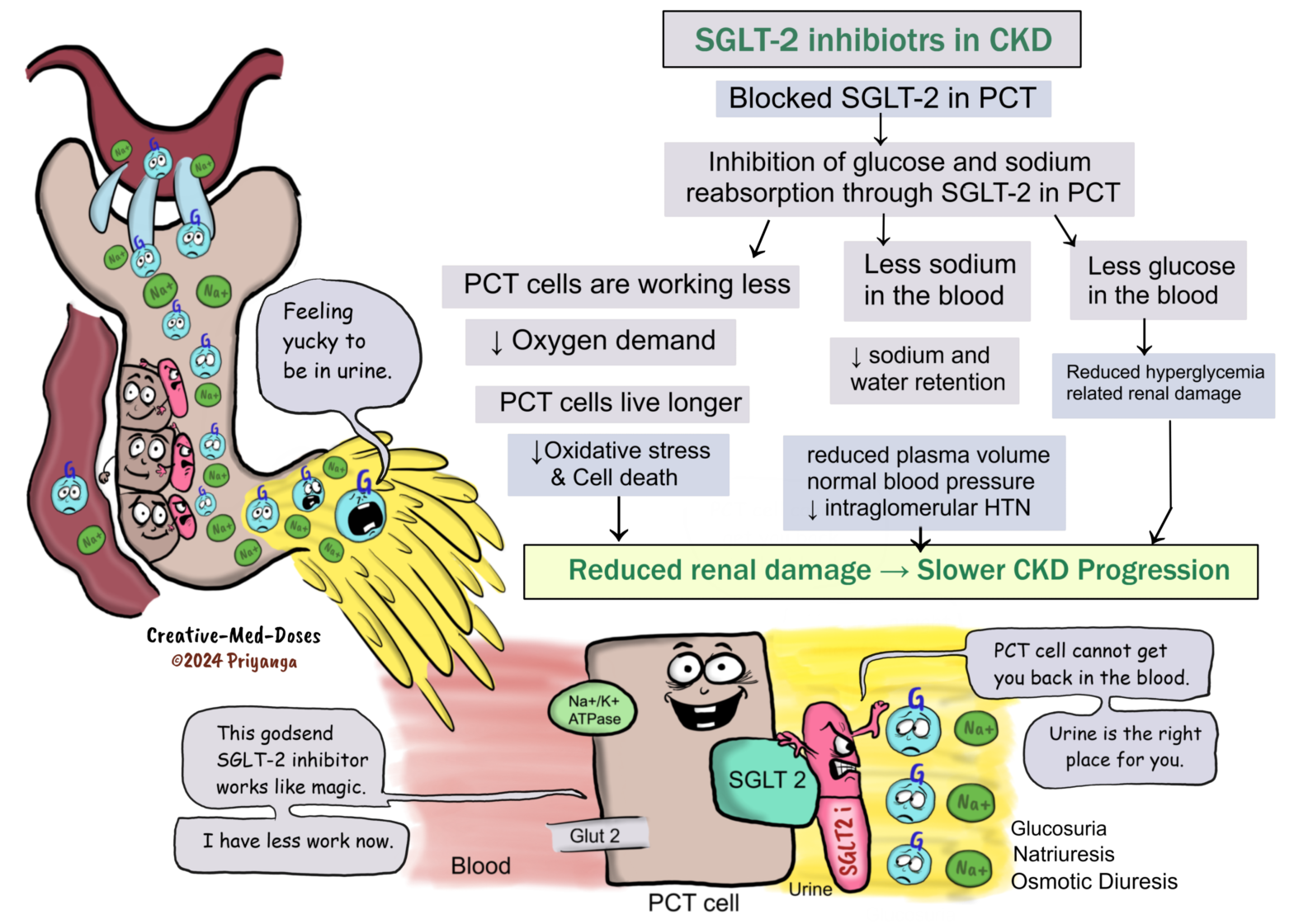
Sodium-glucose cotransporter -2 Inhibitors (SGLT-2 i) are newer drugs in the category of glucose-lowering drugs (antihyperglycemics). The SGLT-2 inhibitors block the SGLT-2 protein in proximal convoluted tubular cells (PCT cells) and inhibit the reabsorption of glucose and sodium, consequently reducing blood glucose and sodium levels.
Nephrogenic systemic fibrosis : (nephrogenic fibrosing dermopathy)

Nephrogenic systemic fibrosis (NSF), as the name implies, is a systemic fibrosis involving multiple organs and tissues due to nephrogenic pathology (chronic/acute renal failure).
Previously, NSF was known as nephrogenic fibrosing dermopathy because of peculiar skin manifestation detected at the time of discovery of this entity. But now it is discovered that it involves other organs and tissues, so Nephrogenic systemic fibrosis is a better term to describe the disease.
Metabolic Acidosis in Chronic Kidney Disease
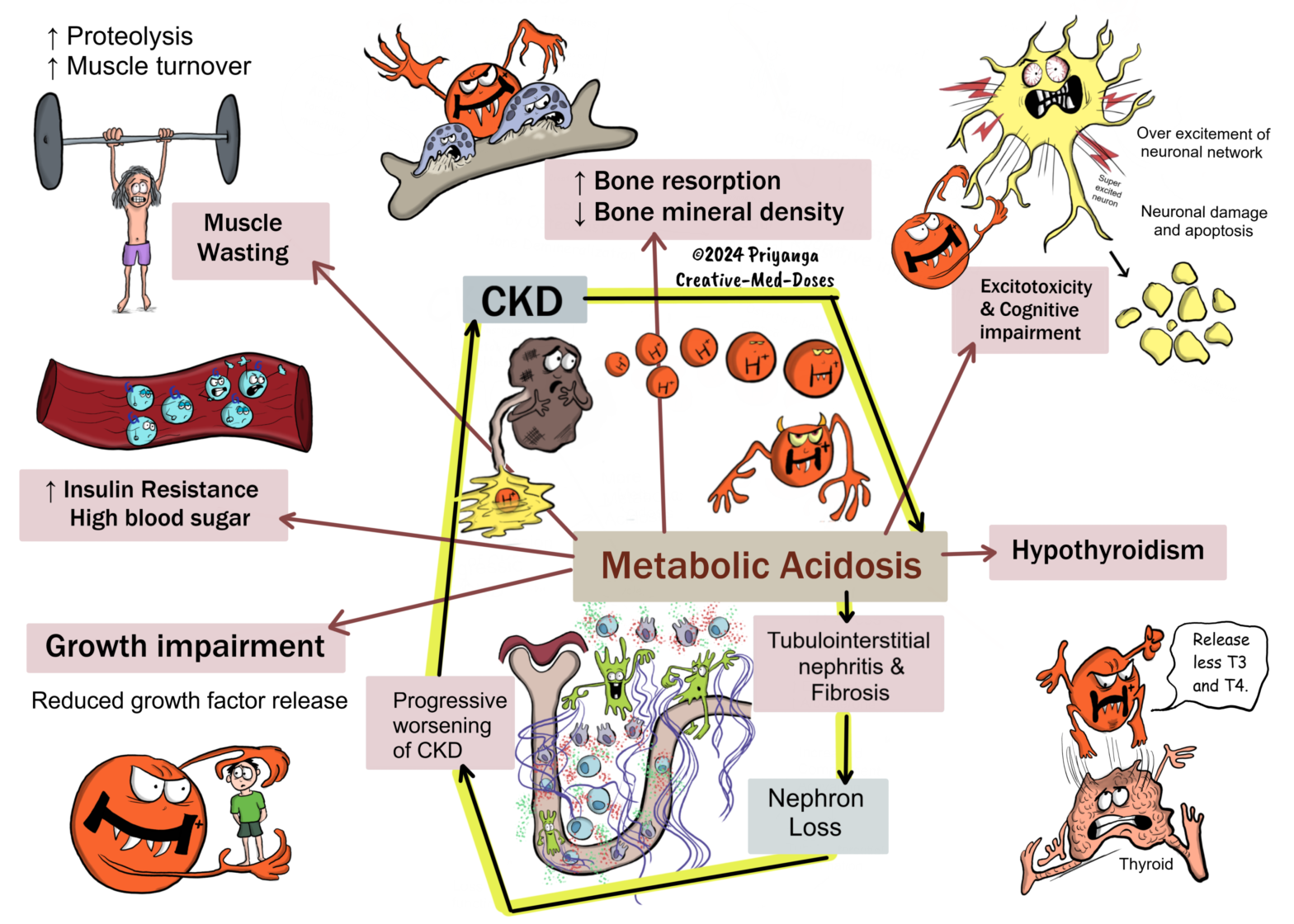
Metabolic acidosis is a condition where the body’s acid load is so high that it starts to affect the normal functioning of the body. It is defined as serum bicarbonate levels < 22 mEq/L (or serum tCO2 levels < 22 mEq/L). The lungs and kidneys play a major role in acid-base maintenance. Serum bicarbonate and serum tCO2 Serum bicarbonate travels through the body in the form of carbon dioxide gas. High serum bicarbonate gives high levels of serum total carbon dioxide (tCO2), and low serum bicarbonate levels give low levels of serum tCO2. That’s why the serum tCO2 is a surrogate assessment marker for serum bicarbonate levels. Normal kidney removes excess acid load by excreting more ammonia and titratable acids. In chronic kidney disease, the kidney cannot excrete excess acid due to a reduced number of functioning nephrons. It leads to the accumulation of acid in the blood and metabolic acidosis. Metabolic acidosis and serum bicarbonate levels The body has a carbonic acid-bicarbonate buffer system. It helps in maintaining the pH balance. The lungs and kidneys play a crucial role in regulating the acid-base balance using this buffer system.
Bronchiectasis
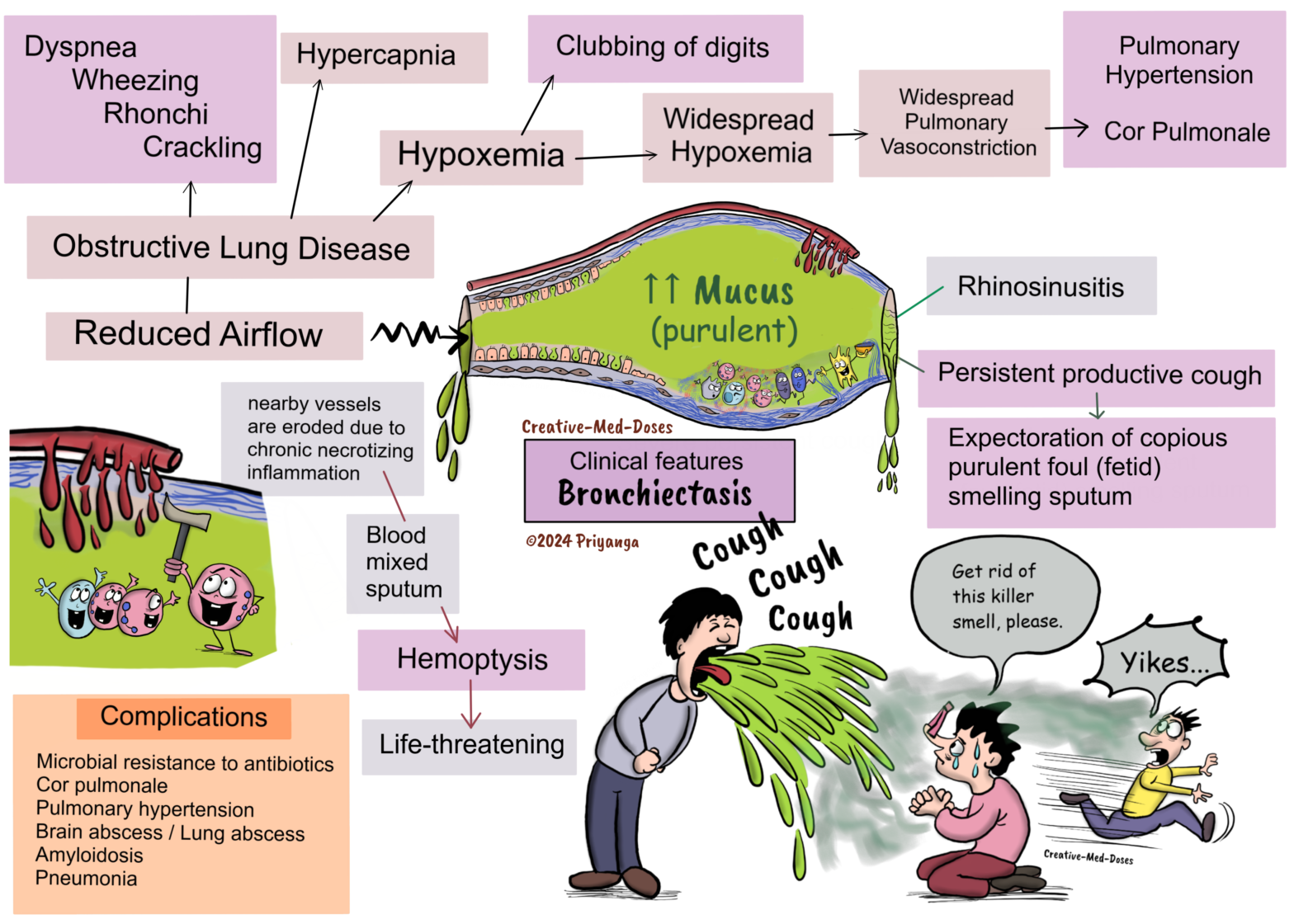
Bronchiectasis is an obstructive lung disease characterized by chronic necrotizing damage of smooth muscles and elastic support tissue of bronchi and bronchioles. It consequently leads to permanent irreversible dilation of bronchi and bronchioles. The involved bronchi and bronchioles are filled with excessive mucus and lead to reduced airflow. The bronchial dilation can be cylindrical (most common), varicose, or cystic.
The lung involvement can be focal or diffused.
• Focal – seen in tumor or foreign body impaction, bronchiectasis is localized to the obstructed lung segment.
• Diffused – seen in infection or immunodeficiency state, widespread bronchiectasis involving the entire lung.
Etiopathogenesis
Chronic necrotizing inflammation of the bronchi, caused by bronchial obstruction/ infection or impaired mucociliary clearance, is central to the pathogenesis of most cases of bronchiectasis.
Obstruction- bronchial obstruction due to tumor (internal/ external), lymphadenopathy, or foreign body
Infection – tuberculosis, staphylococcus aureus, pseudomonas aeruginosa
Impaired Mucociliary Clearance
Cystic fibrosis
Mutation in CFTR gene → Reduced chloride ion secretion in the mucus of respiratory epithelium → Thick and viscid mucus → Cilia cannot move smoothly in thick viscid mucus →Impaired Mucociliary clearance →Reduced removal of pathogens →Superimposed infection→ Chronic necrotizing inflammation → Bronchial wall damage → Permanent bronchial dilation → Bronchiectasis.
Please read more about Cystic Fibrosis – Creative Med Doses
Primary ciliary dyskinesia
Autosomal recessive mutation → Defect in motile cilia production or function → Immotile cilia is unable to sweep mucus → Impaired Mucociliary clearance→ Reduced removal of pathogens → Superimposed infection → Chronic necrotizing inflammation → Bronchial wall damage → Permanent bronchial dilation → Bronchiectasis.
Immotile cilia also lead to infertility and dextrocardia
Kartagener syndrome
• Situs inversus
• Chronic sinusitis
• Bronchiectasis
Read about Kartagener syndrome where cilia doesn’t move – Creative Med Doses
Clinical features
• Persistent productive cough
• Expectoration of copious purulent foul (fetid) smelling sputum
• Tenacious sputum
• Acute exacerbation is associated with sputum changes (increased purulence and volume)
• Hemoptysis (when nearby vessels erode due to chronic necrotizing inflammation)
• Wheezing and crackling on auscultation
• Clubbing of digits due to chronic hypoxemia
Diagnosis
Based on clinical presentation and consistent radiological findings.
Chest X-ray – Shows tram tracks sign (thickened bronchial wall)
High-Resolution Computed Tomography (HRCT)- Imaging modality of choice
• Tram track sign- Bronchial wall thickening (parallel tram lines)
• Tree bud appearance – Dilated and thickened bronchial wall with intraluminal inspissated mucus, pus, and fluid resembling a budding tree.
• Signet ring sign- In bronchiectasis, a cross-sectional area of the airway is at least 1.5 times that of the adjacent vessel, and it appears like a signet ring. The bronchus and artery diameter should be the same size, but in bronchiectasis, the dilated bronchi diameter is more than 1.5 times that of the adjacent artery. It gives a signet ring appearance.
• Lack of bronchial tapering (the presence of tubular structures within 1 cm from the pleural surface)
• Cystic dilation of bronchi
Bronchoscopy is done to rule out foreign body or mass causing airway obstruction, especially in cases with focal bronchiectasis.
Pulmonary function testing for functional assessment of the patients – It can be an obstructive, restrictive, or normal pattern.
Polycythemia vera
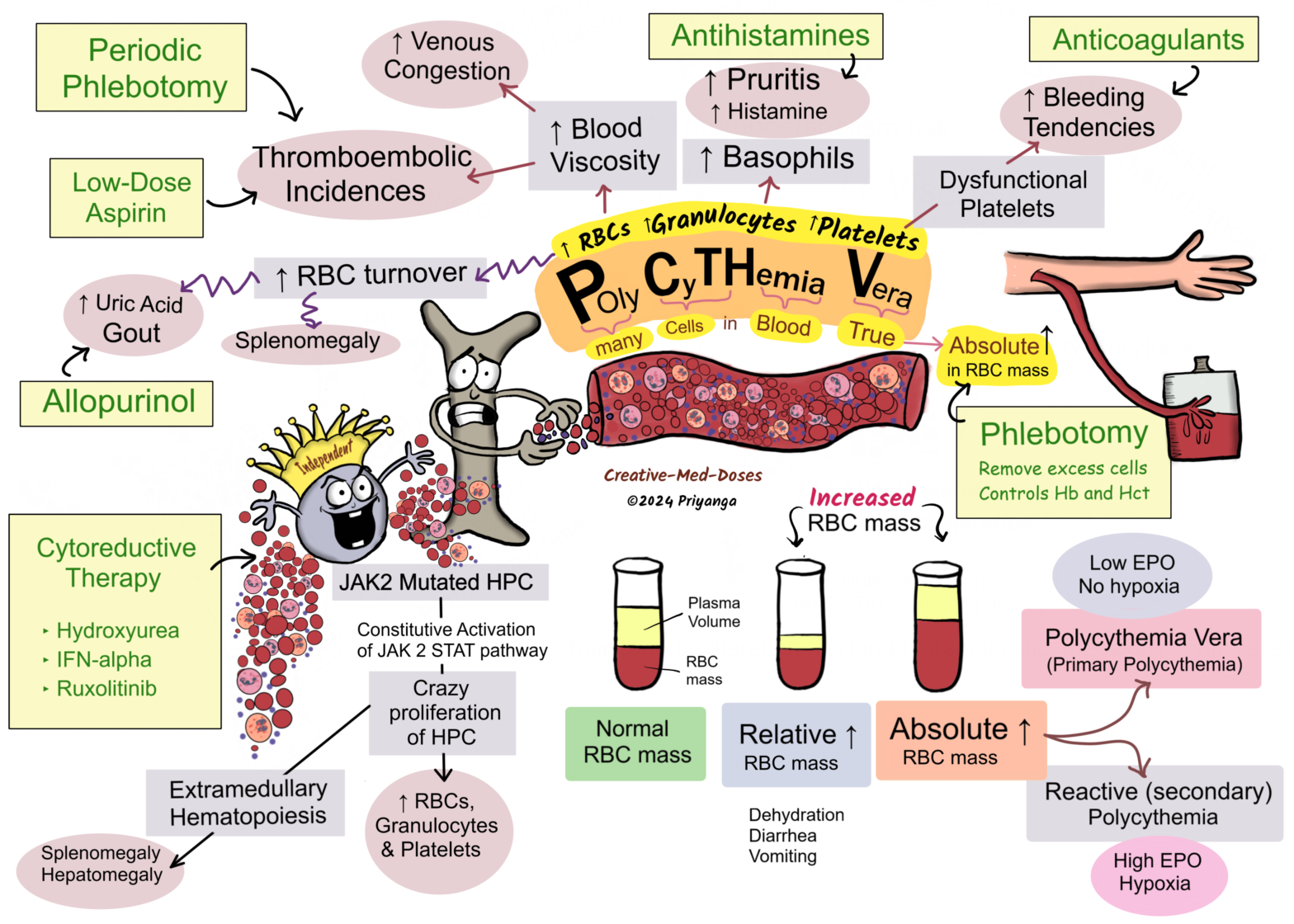
Polycythemia vera (PV) is a chronic myeloproliferative disorder with trilineage proliferation (erythroid, granulocytic, megakaryocytic).
It leads to an increased number of RBCs, granulocytes (Neutrophils, basophils, and eosinophils), and Platelets.
It is associated with an absolute increase in RBC mass.
PV must be differentiated from relative polycythemia, which results from hemoconcentration due to diarrhea/ vomiting. Polycythemia vera, in contrast to reactive polycythemia, is associated with low levels of serum erythropoietin (EPO).
Genetics
Most cases (95%) are associated with JAK2 V617F mutation or JAK2 axon 12 mutation on the short arm of the 9th chromosome.
JAK2 617F mutation is also seen in nearly 50% of cases of –
• Essential thrombocythemia (ET)
• Primary myelofibrosis (PMF)
Pathogenesis
JAK2 V617F point mutation or JAK2 axon 12 mutation on the short arm of the 9th chromosome → constitutive activation on JAK2/STAT signaling pathway → sharply lowers the dependence of hematopoietic cells on growth factors for growth and survival → Dysregulated activation of JAK-STAT signaling pathway → Proliferation and differentiation of Hematopoietic progenitor cells → Too much increase in RBC, Granulocytes, and Platelets (panmyelosis) → most clinical signs and symptoms are related to an absolute increase in red cell mass → Polycythemia Vera (Primary polycythemia)