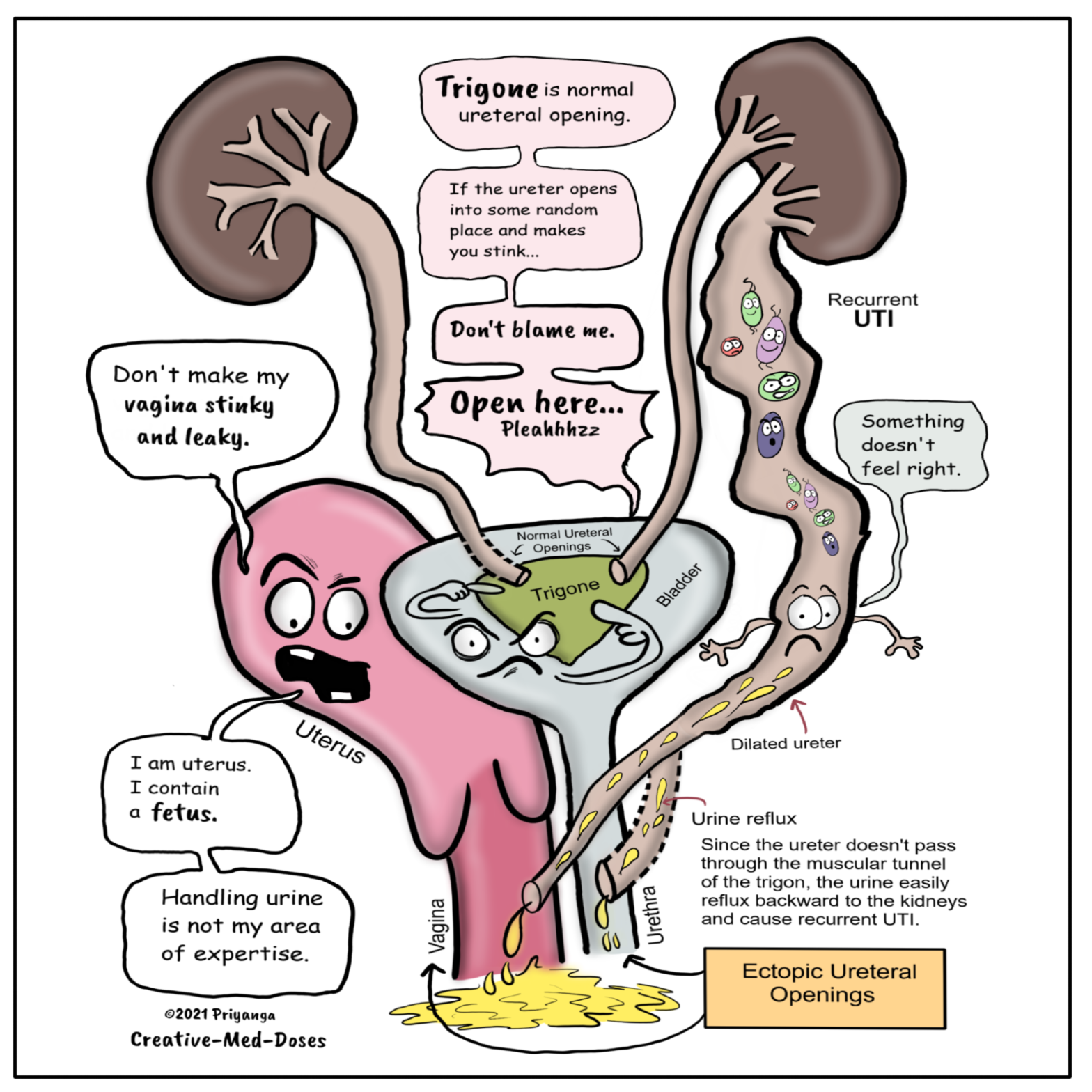
Ureterocele is a congenital dilation of the intravesicular portion of a ureter. In this anomaly, the part of the distal ureter which enters the bladder balloons at its opening into the bladder, forming a sac-like pouch. It appears balloon-shaped on cystoscopy. Most ureteroceles get prenatal diagnosis while doing routine antenatal Ultrasound. The ureterocele causes prenatal hydronephrosis. And, in the child, it causes frequent urinary tract infections because it is associated with vesicoureteral reflux.
It can be of two types –
A ureterocele is a congenital dilation of the intravesicular portion of a ureter. In this anomaly, the part of the distal ureter which enters the bladder balloons at its opening into the bladder, forming a sac-like pouch. This sac-like enlargement usually interferes with the flow of urine.
Post Type Archives: Topics
Complement-dependent cytotoxicity (CDC) crossmatch
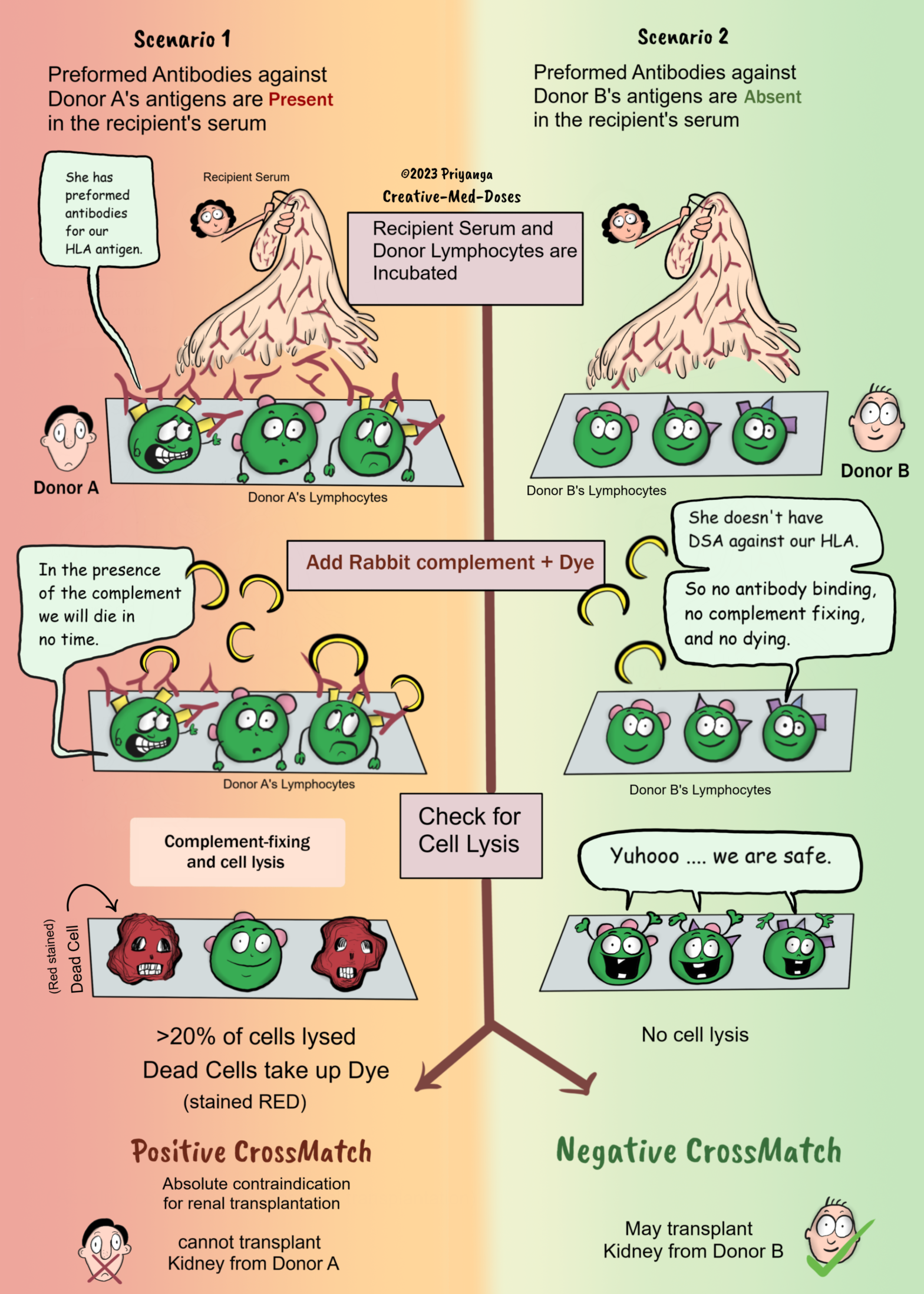
Complement-dependent cytotoxicity (CDC) crossmatch is the most commonly performed test to determine whether one should proceed with transplantation.
In the complement-dependent cytotoxicity (CDC) crossmatch test donor’s lymphocytes from blood or lymphoid tissue are incubated with recipient serum, followed by mixing rabbit complement and dyes to distinguish dead from living donor cells.
The cell membrane is not intact in lysed dead cell, so it will take up the dye and stain red (or whatever color the dye contains).
If the recipient’s serum has preformed antibodies against the donor’s antigens, there will be cell lysis after adding complement, and the dead cells will take up the dye. It is a positive crossmatch if there is cell lysis of more than 20 percent of donor cells (lymphocytes). Avoid transplantation from such donors. Positive CDC crossmatch is an absolute contraindication for renal transplantation.
It is a negative crossmatch if the donor’s lymphocytes show no or minimal cell lysis. One may go with transplantation because the donor and recipient are compatible, and the chances of early graft rejection are low.
Tubulointerstitial nephritis
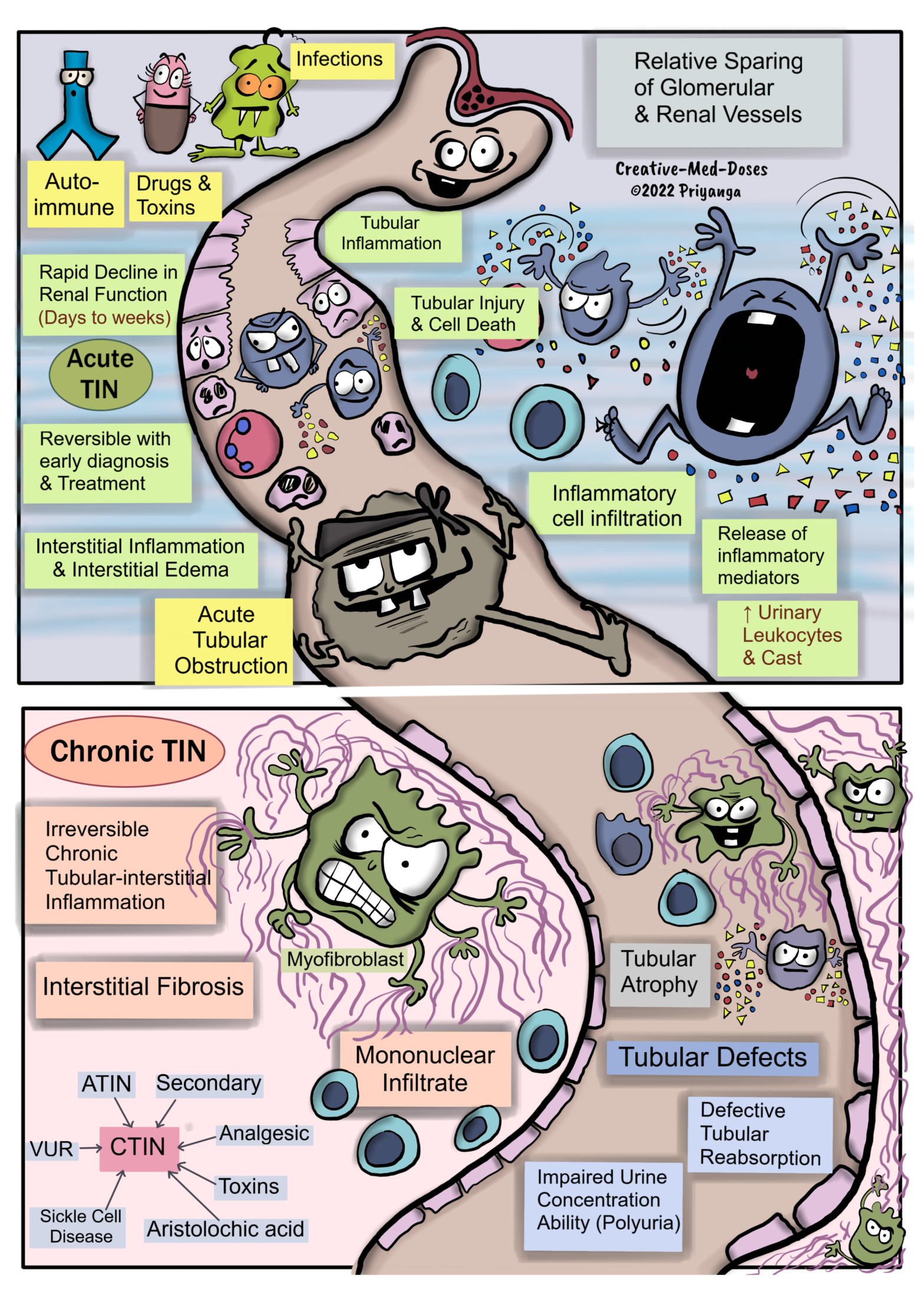
Tubulointerstitial nephritis (TIN)
Introduction
Tubulointerstitial diseases can be of two types- Primary and Secondary
Secondary tubulointerstitial disease: Inflammation or fibrosis of the renal interstitium and atrophy of the tubular compartment following the glomeruli or vascular diseases.
Primary Tubulointerstitial Nephritis (TIN)- primarily affects the tubules and interstitium, with relative sparing of the glomeruli and renal vessels. Glomerulus and renal vessels get involved in the later stages of the disease.
Primary Tubulointerstitial nephritis can be acute and chronic tubulointerstitial nephritis (TIN).
Acute Tubulointerstitial Nephritis (ATIN)-Tubulo-interstitial inflammation causes a rapid decline in renal function within days to weeks. It is reversible with early diagnosis and prompt treatment.
Causes and Pathogenesis of ATIN
Drug-induced TIN
Following drugs are the most common etiological factor of Drug-induced ATIN-
• Penicillin
• Rifampicin
• Diuretics
• Proton pump inhibitors
• NSAIDs
• Sulfa Drugs
• Phenytoin
Tubular secretion of the drug→ Drug act as a HAPTEN →Drug binds with the cytoplasmic or extracellular components of Tubular Cells →Drug becomes IMMUNOGENIC → Immune-mediated tubular-interstitial damage (Type I hypersensitivity or Type IV hypersensitivity).
Type I hypersensitivity
IgE mediated Tubular and Interstitial damage
Classical Triad of Drug-Induced TIN
• Eosinophilia
• Rashes
• Fever
Type IV hypersensitivity – T Cell-mediated Tubular and interstitial damage
Autoimmune Diseases
Autoantibodies and immune complexes lead to Immune-mediated tubular-interstitial damage.
Most common autoimmune diseases associated with Acute TIN-
• Tubulointerstitial Nephritis and Uveitis Syndrome (TINU)
• Sjogren Syndrome
• SLE
Acute Tubular Obstruction
Light Chain Cast Nephropathy (LCCN), also known as Myeloma Kidney/ Crystal Nephropathy (uric acid/medications) →Acute tubular obstruction →Tubular injury and inflammation → Tubulointerstitial nephritis and acute tubular necrosis if left untreated →Acute Renal Failure.
Microbial Infection
Microbial infection (bacteria/virus/fungi) → stimulates tubulointerstitial inflammation →Acute TIN.
The microbial infection causes ATIN only in Preexisting Tubular obstruction /Vesicoureteral Reflux or Immunocompromised cases.
Multiple Myeloma associated renal injury
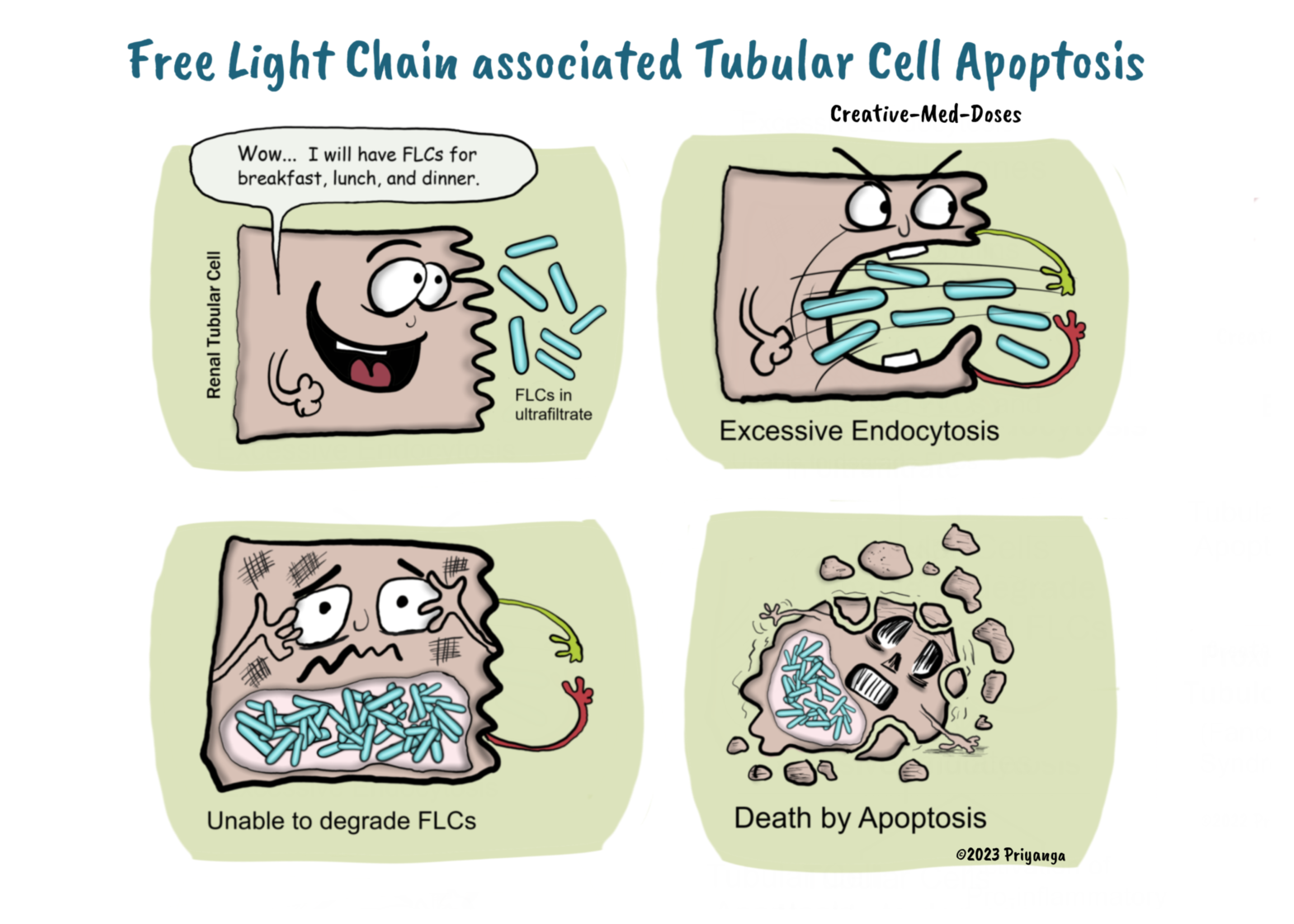
Multiple Myeloma (MM) is a plasma cell dyscrasia associated with malignant plasma cells producing excessive monoclonal proteins (immunoglobulin and free light chain).
Renal injury is commonly associated with MM and has very high mortality rates. More than 50% of cases of MM present with renal involvement.
Renal Papillary Necrosis (RPN)
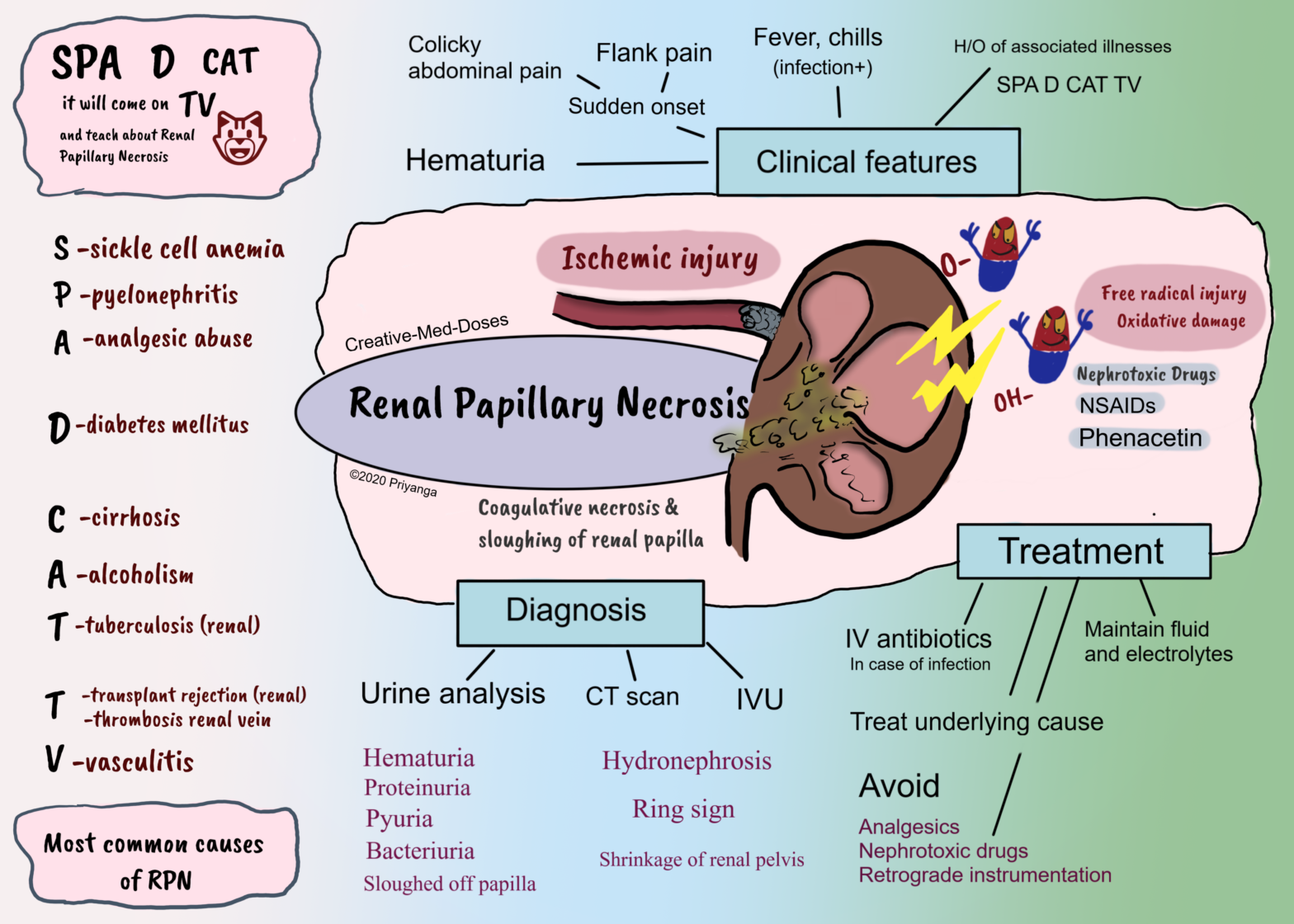
Renal Papillary Necrosis (RPN)
Renal papillary necrosis (RPN) is coagulative necrosis and sloughing off of renal papilla after ischemia or oxidative damage.
The renal papilla is the rounded apex of each medullary pyramid. Each papilla represents the confluence of the collecting ducts from each nephron within that pyramid.
The papillary tip receives only a marginal supply of blood from small-caliber vessels. The already tenuous vascular supply makes renal papilla vulnerable to ischemia and necrosis.
Etiopathogenesis
Reduced blood supply to renal papilla → Ischemia →coagulative necrosis and sloughing of renal papilla → necrosed tissue get infected or calcified → or it can slough off and obstruct urinary tract. A large amount of necrosed tissue causes acute urinary tract obstruction and renal failure.
Nephrotoxic drugs can cause renal papillary necrosis by toxic effects and oxidative damage to cells of renal papilla.
Following are the most common causes associated with RPN-
SPA D CAT because it will come on TV and teach Renal Papillary Necrosis
SPA S- sickle cell anemia P- pyelonephritis A- analgesic abuse
D-diabetes Mellitus
C-cirrhosis A-alcoholism T-tuberculosis
T- transplant rejection, thrombosis of renal vein V-vasculitis
Barrett’s esophagus
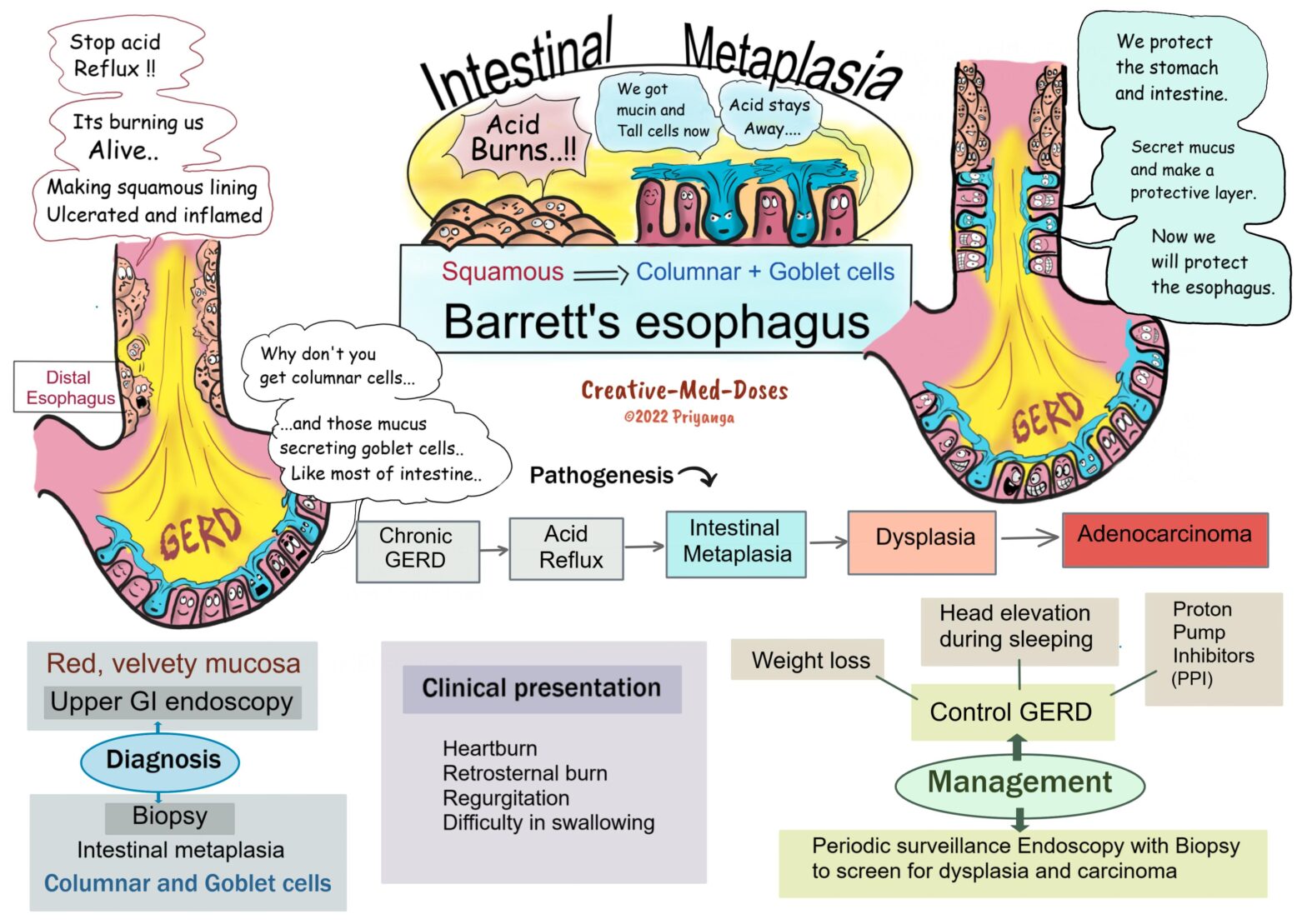
Barrett’s esophagus is associated with chronic gastroesophageal reflux (GERD) and reflux esophagitis. Long-term acid exposure induces metaplastic changes in distal esophageal mucosa. And the esophageal squamous epithelium is replaced by columnar and goblet cells (intestinal metaplasia).
Pathogenesis
Chronic GERD → Increased acid exposure → Esophageal mucosal injury and squamous epithelial damage → Acute and chronic inflammatory changes → Reflux esophagitis → Recovery from damaged area involves metaplastic changes (Barrett’s esophagus) → Esophageal stem cells develop intestinal metaplasia (columnar and goblet cells) → Metaplasia may lead to dysplasia → Adenocarcinoma of the esophagus
Associated Risk Factors
• GERD
• Obesity
• Men > Women
Clinical Presentation of Barrett’s esophagus
• Heartburn
• Regurgitation
• Difficulty in swallowing
Diagnosis
History of chronic GERD and clinical presentation
Upper GI Endoscopy
Endoscopic examination shows tongues and patches of red, velvety mucosa extending upwards from the gastroesophageal junction. The normal esophageal mucosa looks smooth, pale, and pearly white.
Biopsy
Gastric and intestinal metaplasia is present in mucosa above the gastroesophageal junction. The presence of goblet cells with mucous vacuoles is defining feature of intestinal metaplasia. Goblet cells stain pale blue by H&E stain.
Focal Segmental Glomerulosclerosis (FSGS)
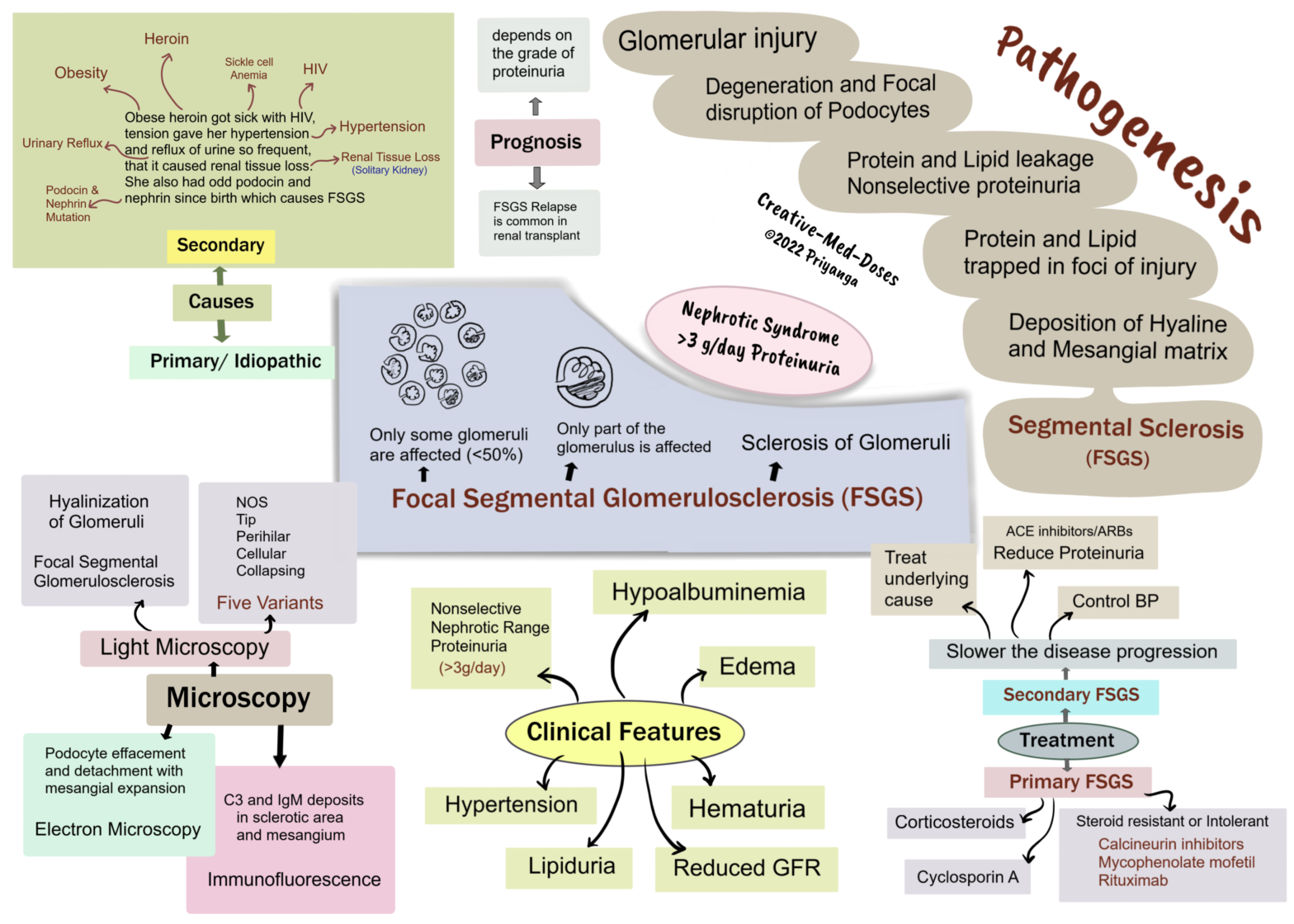
Focal segmental glomerulosclerosis (FSGS) is characterized by sclerosis of <50% of glomeruli (Focal). And sclerosis involves only a part of each affected glomerulus(segmental).
The most common Primary glomerular lesions that lead to the nephrotic syndrome are focal segmental glomerulosclerosis (in adults) and minimal-change disease (in children).
Types and causes
FSGS can be primary and secondary. This distinction has both prognostic and therapeutic implications.
Primary FSGS (idiopathic) - approximately 20% to 30% of all cases of nephrotic syndrome.
Secondary FSGS may be associated with the following conditions -
• HIV infection
• Heroin abuse
• Obesity
• Sickle cell disease
• Secondary to other forms of GN (e.g., IgA nephropathy, hypertensive nephropathy, reflux nephropathy)
• As a maladaptation to nephron loss due to chronic kidney disease or congenital malformations
• Inherited forms are associated with mutations in cytoskeletal proteins nephrin and podocin. Nephrin and podocin are essential for the integrity of podocytes.
Pathogenesis
Multiple factors/defects → Glomerular injury → Degeneration and Focal disruption of Podocytes →Decreased Glomerular barrier integrity →Protein and Lipid leakage →Nonselective proteinuria→ Nephrotic range >3g/day→ Protein and Lipid trapped in foci of injury →Deposition of Hyaline and Mesangial matrix→ Sclerosis of injured part of glomerulus →Segmental Sclerosis→ Focal Segmental Glomerulosclerosis.
Adaptive FSGS
Hypertension-related renal damage→ Loss of autoregulation of renal blood flow at the afferent arteriole
→Transmission of elevated pressures to an unprotected glomerulus →Leading to hyperfiltration and hypertrophy→ Increased fibrosis and sclerosis→ Eventual focal segmental glomerular sclerosis.
Morphology
Light microscopy- focal segmental sclerosis and hyalinization of glomeruli. Juxtamedullary nephrons are affected first, and inadequate sampling may miss focal lesions.
Histological Variants- FSGS is classified into five variants:
Not Otherwise Specified (NOS)- It is the most common variant of FSGS
Tip – the segmental lesion involves a tubular pole of the glomerulus.
Perihilar – sclerosis and hyalinization at the vascular pole of the glomerulus. It is present in cases with hyperfiltration and adaptive response where glomerular pressure is very high.
Cellular- hypercellular glomerulus with endocapillary and epithelial hyperplasia is characteristic of this variant.
Collapsing- is characterized by the collapse of the glomerular tuft and epithelial cell hyperplasia. It has the lowest rate of remission and the worst prognosis.
Immunofluorescence – C3 and IgM deposits in sclerotic area and mesangium.
Electron Microscopy –shows podocyte effacement and detachment with mesangial expansion.
C3 Glomerulopathy
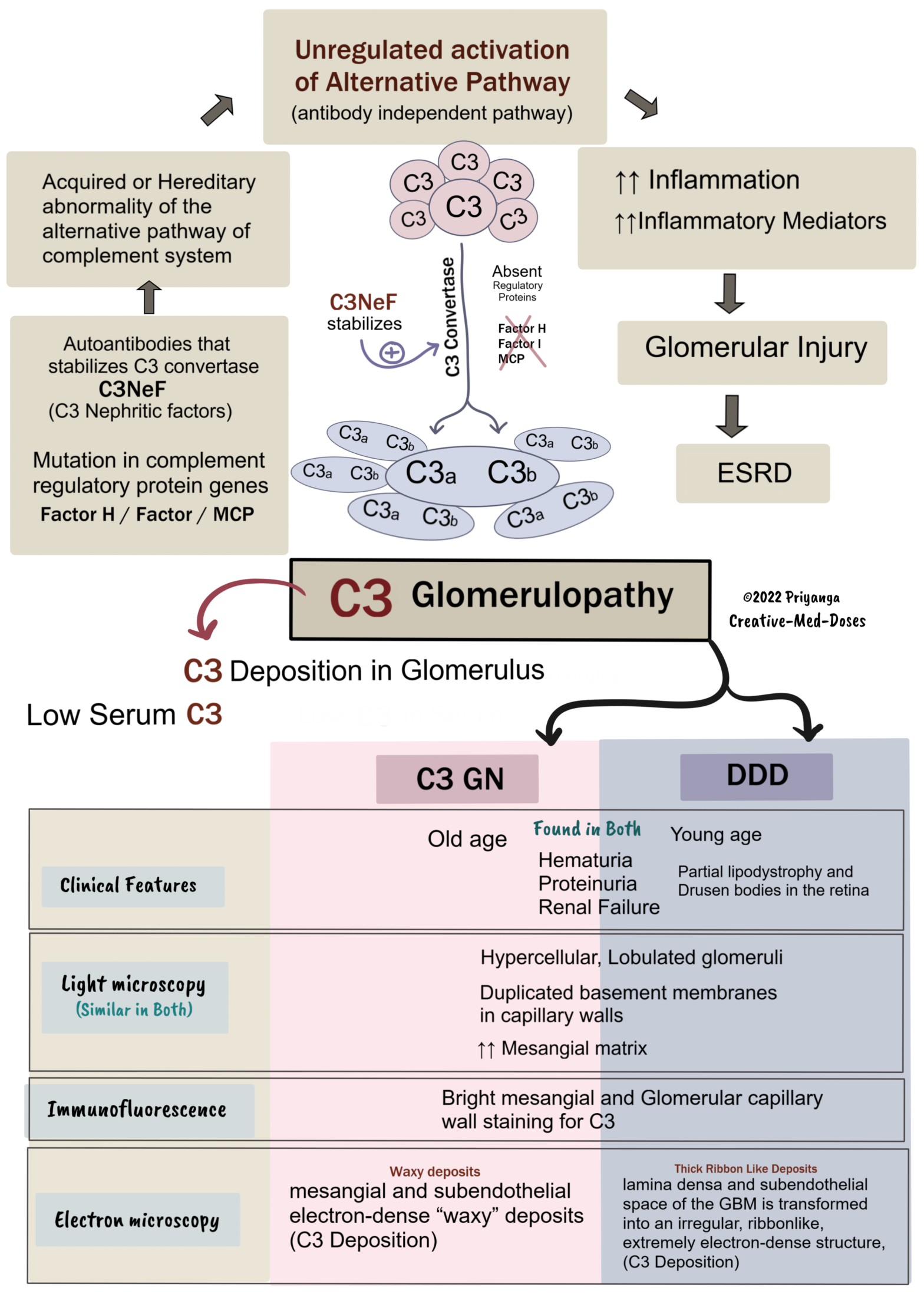
C3 glomerulopathy is a broad term encompassing-
• dense deposit disease (DDD), former MPGN type II
• C3 glomerulonephritis
Both have low serum C3 levels (hypocomplementemia)
C3 glomerulonephritis comprises examples of MPGN types I and III, in which immunofluorescence reveals predominant C3 deposits.
Light microscopy
Both C3GN and DDD have similar Light microscopy findings.
• Hypercellular, Lobulated glomeruli
• Duplicated basement membranes in capillary walls
• ↑↑ Mesangial matrix
Immunofluorescence – Similar in both C3GN and DDD
Bright mesangial and glomerular capillary wall staining for C3. Immunoglobulins (IgG) are absent.
Electron Microscopy – (different electron dense pattern)
C3GN
Mesangial and subendothelial electron-dense “waxy” deposits of C3.
DDD
lamina densa and subendothelial space of the GBM are transformed into an irregular, ribbonlike, and extremely electron-dense structure.
Bartter syndrome
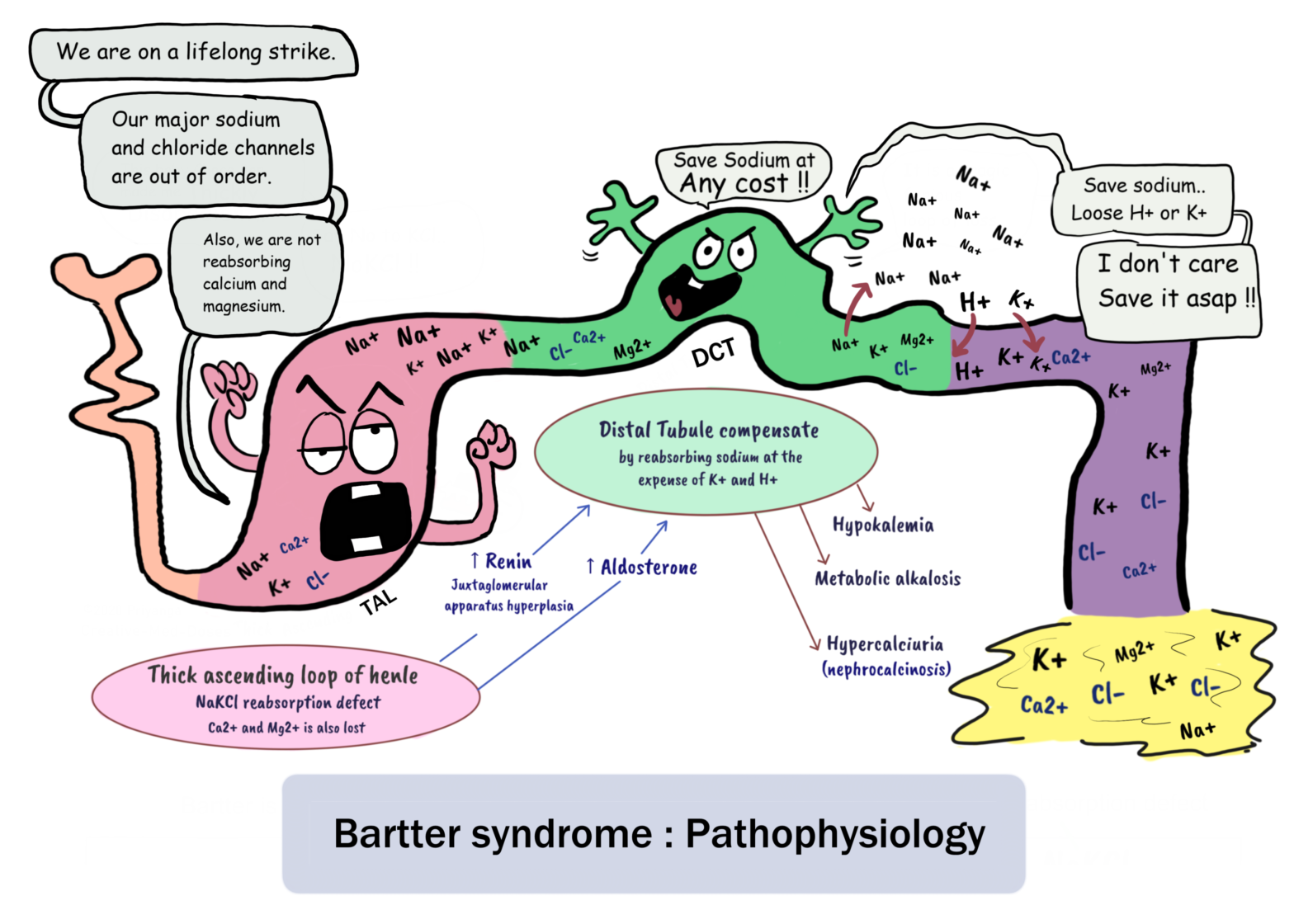
Bartter syndrome is an autosomal recessive salt-wasting renal tubular disorder. It is characterized by hypokalemia, hypochloremia, metabolic alkalosis, and high renin with normal/low blood pressure. This disorder mimics chronic loop diuretic (furosemide).
It results from mutations in various genes that affect the function of ion channels and transporters that mediate transepithelial salt reabsorption in the thick ascending limb of the loop of Henle.
Etiology
Impairment in the sodium-potassium-chloride cotransporter (NKCC2) or the potassium channel (ROMK) affects the transport of sodium, potassium, and chloride in the thick ascending limb of the loop of Henle (TALH) and leads to Bartter syndrome.
Types of Bartter syndrome:
• Type I results from mutations in the sodium chloride/potassium chloride cotransporter gene (NKCC2)
• Type II results from mutations in the ROMK gene.
• Type III results from mutations in the chloride channel gene (CLC-Kb)
• Type IV results from the loss-of-function mutations in gene encoding barttin. Barttin is required for both chloride channels ClC-Kb and ClC-Ka to function.
• Type V results from mutations in extracellular calcium ion-sensing receptors and in the genes that encode the chloride channel subunits, ClC-Ka and ClC-Kb.
Pathophysiology
Bartter syndrome is a renal tubular salt-wasting disorder where a mutation in various sodium and chloride transporting channels genes leads to impaired reabsorption of sodium and chloride in the thick ascending limb of the loop of Henle.
Impaired chloride reabsorption in the thick ascending limb of the loop of Henle results in malabsorption of calcium thick ascending limb (TAL). Under normal conditions, calcium and magnesium are absorbed paracellularly under the influence of a positive charge in the lumen due to the reabsorption of negatively charged chloride ions. Increased calcium loss in urine increases the chances of Osteoporosis and Nephrocalcinosis.
Increased Distal convoluted tubular delivery of sodium, calcium, and magnesium → increased renin through JG apparatus →activation of Renin angiotensin aldosterone system →increased sodium reabsorption in DCT in exchange of potassium and hydrogen ions→ hypokalemia and metabolic alkalosis
Excessive salt and water loss →volume depletion →activation of the renin-angiotensin-aldosterone system (RAAS)
→secondary hyperaldosteronism →Long-term stimulation causes hyperplasia of the juxtaglomerular apparatus →high renin levels with normal blood pressure
Bartter Syndrome: Clinical Presentation
Signs and symptoms of Bartter syndrome are due to electrolyte imbalance, water loss, and its consequences. It mimics long-term ingestion of loop diuretics (furosemide).
In Utero
• Polyhydramnios
• Cord prolapse
• Preterm delivery
• Placental abruption
Infants
• Polyuria
• Polydipsia
• Severe dehydration
• Failure to thrive
Children and adults
• Salt craving
• Polyuria
• Polydipsia
• Normal to low blood pressure
• Failure to thrive
• Vomiting
• Abnormal facies – prominent forehead, large eyes, protruding ears, drooping mouth
• Sensorineural hearing loss
• Cardiac arrhythmias and sudden cardiac death due to electrolyte imbalances
• Osteoporosis due to calcium loss in urine
Treatment
• Tubular defects in Bartter syndrome cannot be corrected, Kidney Transplantation is the only cure.
Management goals
• Intermittent amniocentesis (draining excess amniotic fluid) to treat polyhydramnios
• Normalize potassium levels
• Treat metabolic alkalosis
• Treat and prevent dehydration – saline infusion
Pulmonary Hypertension
Pulmonary hypertension is defined as an elevation in mean pulmonary artery pressure (PAP) > 20 mm Hg or more at rest. Normal PAP levels are 8-20 mmHg.
Hemodynamic definition PH vs PAH
Pulmonary hypertension (PH) = mPAP > 20 mm of Hg at rest
Pulmonary Arterial Hypertension (PAH) has mPAP of >20 mm of Hg at rest but it has Pulmonary capillary wedge pressure (PCWP) <15 mm of Hg (normal PCWP).
Right heart catheterization is the gold standard test for pulmonary artery pressure measurement.
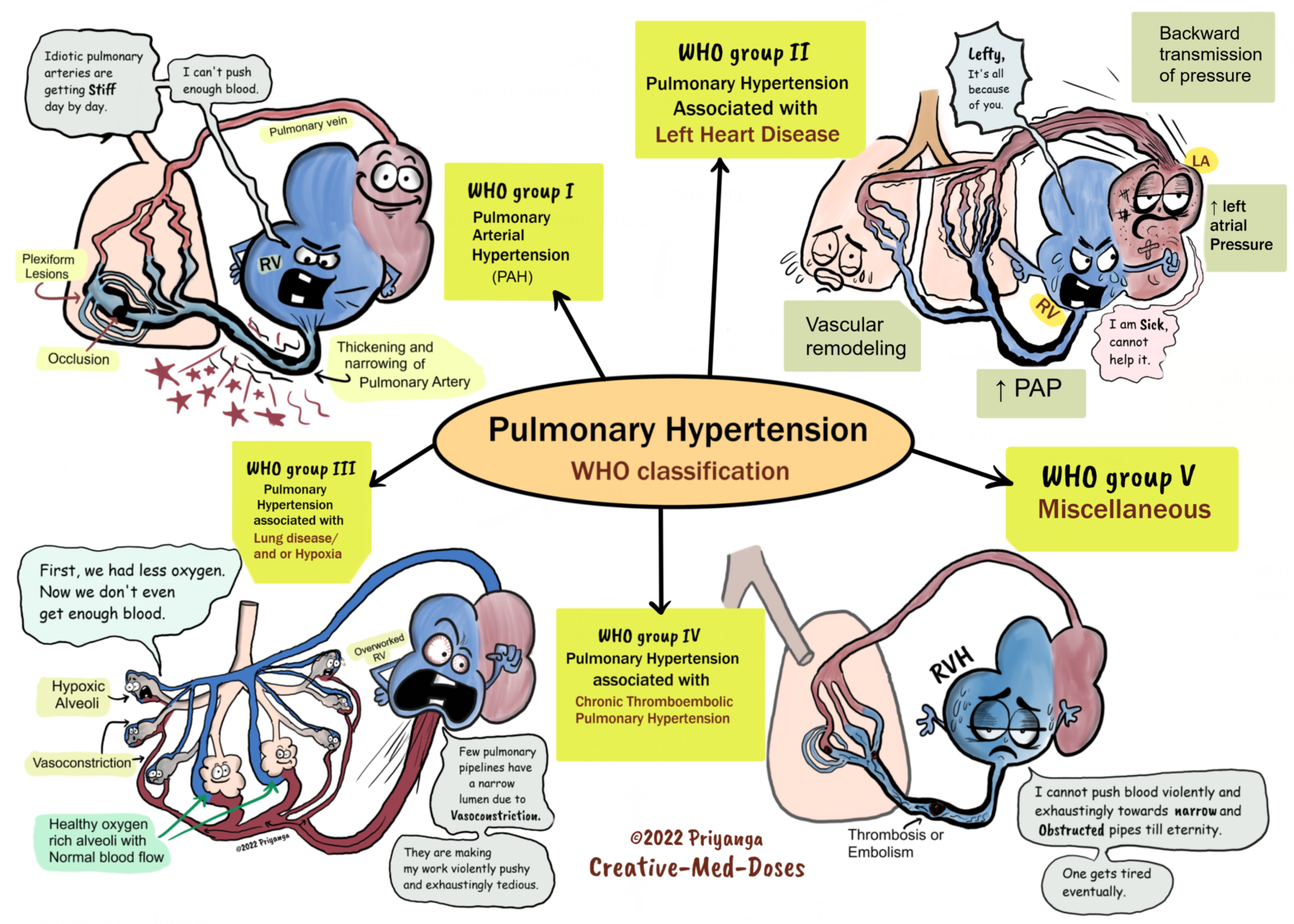
Classification
Pulmonary hypertension is classified into five groups by the World Health Organization (WHO)-
1. Pulmonary arterial hypertension (PAH) is associated with heritable forms of pulmonary hypertension and diseases that cause pulmonary hypertension by affecting small pulmonary muscular arterioles. These include connective tissue diseases, human immunodeficiency virus, and congenital heart disease with a left to right shunt
2. Pulmonary hypertension due to left-sided heart disease, including systolic and diastolic dysfunction and valvular disease
3. Pulmonary hypertension due to lung diseases and/or hypoxia, including COPD and interstitial lung disease
4. Chronic thromboembolic pulmonary hypertension (CTEPH)
5. Pulmonary hypertension with unclear or multifactorial mechanisms