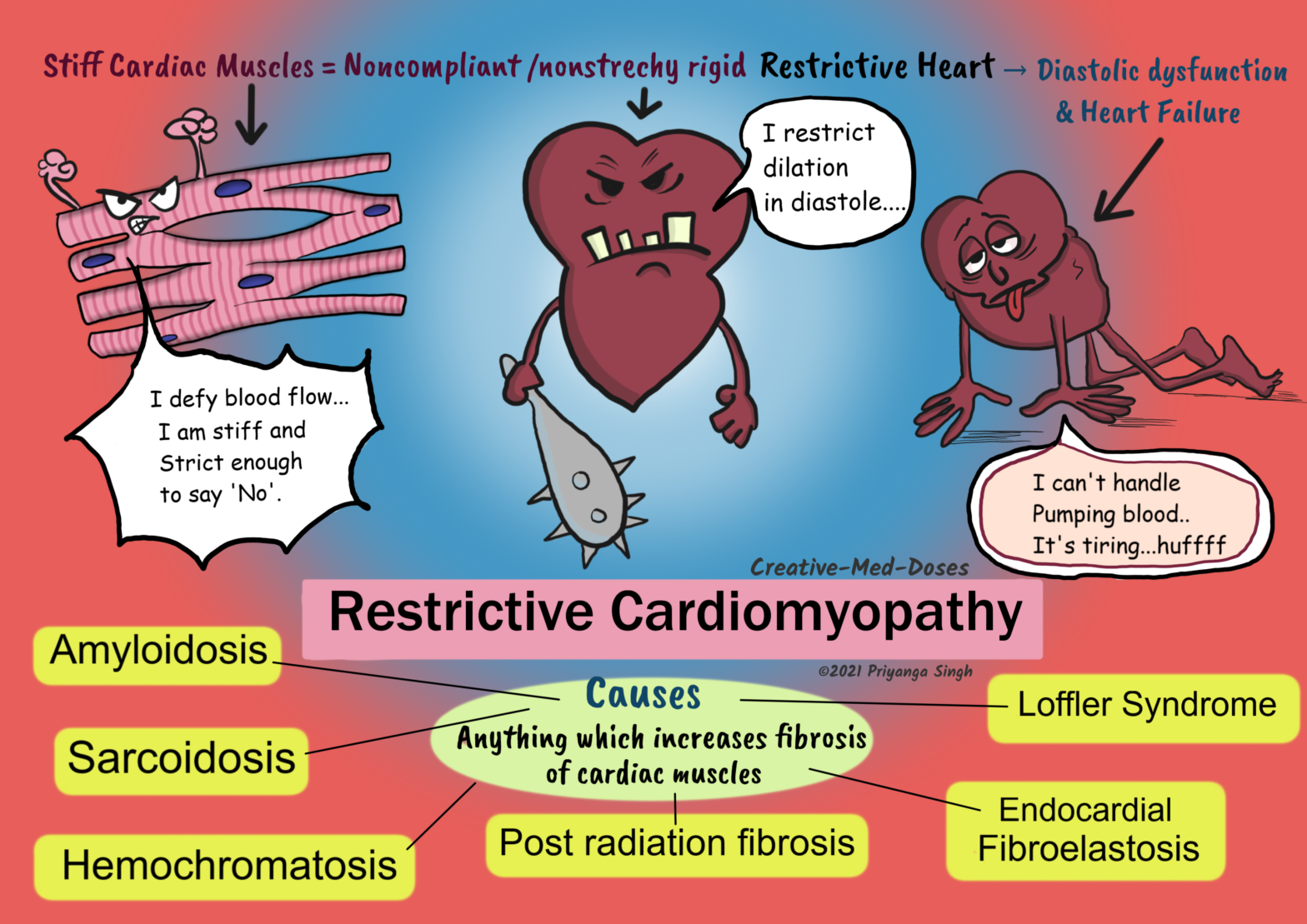
Restrictive cardiomyopathy is a primary disease of ventricular compliance. It causes impaired ventricular filling during diastole because the ventricular wall is stiffer and noncompliant.
Causes
Idiopathic
Amyloidosis- deposition of insoluble β-pleated sheets causes stiff ventricular muscles. Some immunoglobulin light chains are directly cardiotoxic and can induce myocardial dysfunction.
Hemochromatosis- iron deposition can induce free radicle injury and fibrosis of cardiac muscles. It is most commonly associated with dilated cardiomyopathy but can also cause restrictive cardiomyopathy.
Sarcoidosis – granuloma formation, and chronic inflammatory reaction in cardiac muscles cause fibrosis and stiffening of heart muscles
Scleroderma
Post radiation fibrosis – radiation exposure is associated with free radicle formation and fibrosis.
Loffler Syndrome- It has peripheral hyper-eosinophilia, and eosinophilic tissue infiltrates. The release of eosinophil granule contents causes endocardial and myocardial necrosis. It causes the scarring, and layering of the endocardium by thrombus. The thrombus organization leads to stiffened and noncompliant ventricles.
Endomyocardial Fibrosis- there is dense diffuse fibrosis of the ventricular endocardium and subendocardium. It involves the tricuspid and mitral valves. The fibrous tissue markedly diminishes the volume and compliance. Endomyocardial fibrosis is associated with nutritional deficiencies and inflammation related to helminthic infections.
Malignancy
Hepatitis B is a virus which attacks the liver and can cause acute or chronic disease. It is associated with hepatocellular carcinoma. All Hepatitis viruses are hepatophilic viruses (they have an affinity for liver) and that is why they cause liver damage. Transmission Perinatal transmission In highly endemic areas, hepatitis B is most commonly spread […]
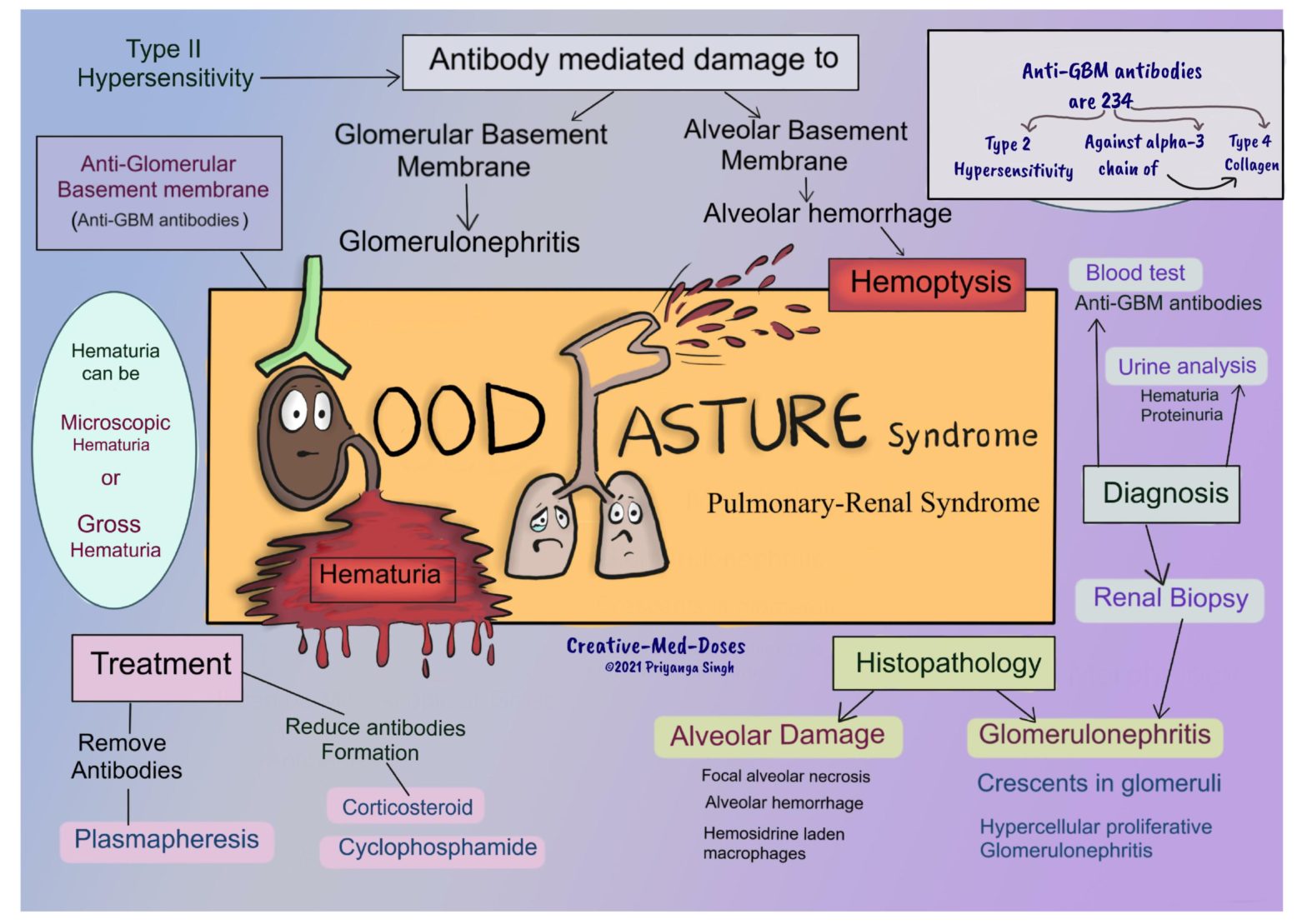
Goodpasture syndrome is an anti-glomerular basement membrane (anti-GBM) disease. It is an autoimmune disease characterized by damage to the alveolar and renal glomerular basement membranes by the anti-GMB antibody. It is a pulmonary-renal syndrome.Goodpasture syndrome characteristically has -• Pulmonary hemorrhage – hemoptysis• Glomerulonephritis-hematuria• circulating anti-glomerular basement membrane antibodies
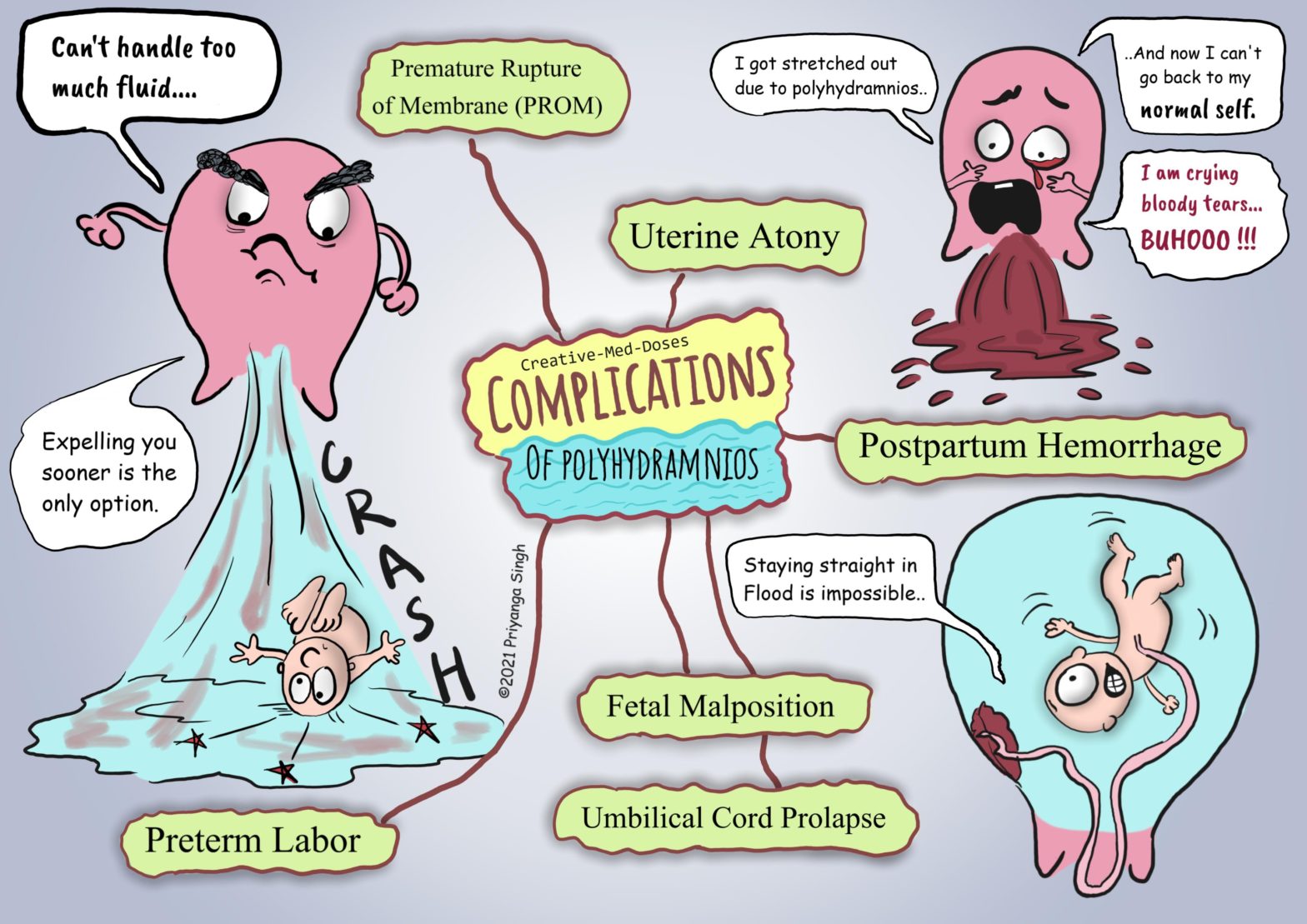
Polyhydramnios is a condition where amniotic fluid exceeds 1.5-2 litres in volume, and an amniotic fluid index (AFI) is greater than 20-25.
Etiology
Excess in amniotic fluid can be due to
Decreased resorption (decreased fetal swallowing) –
The gastrointestinal obstruction associated abnormalities like intestinal atresia, tracheoesophageal fistula, or anencephaly cause reduced swallowing of amniotic fluid by the fetus. It leads to excessive amniotic fluid in the uterine cavity.
Increased production (excess fetal urination)- Fetal polyuria, as in maternal diabetes, leads to increased amniotic fluid.
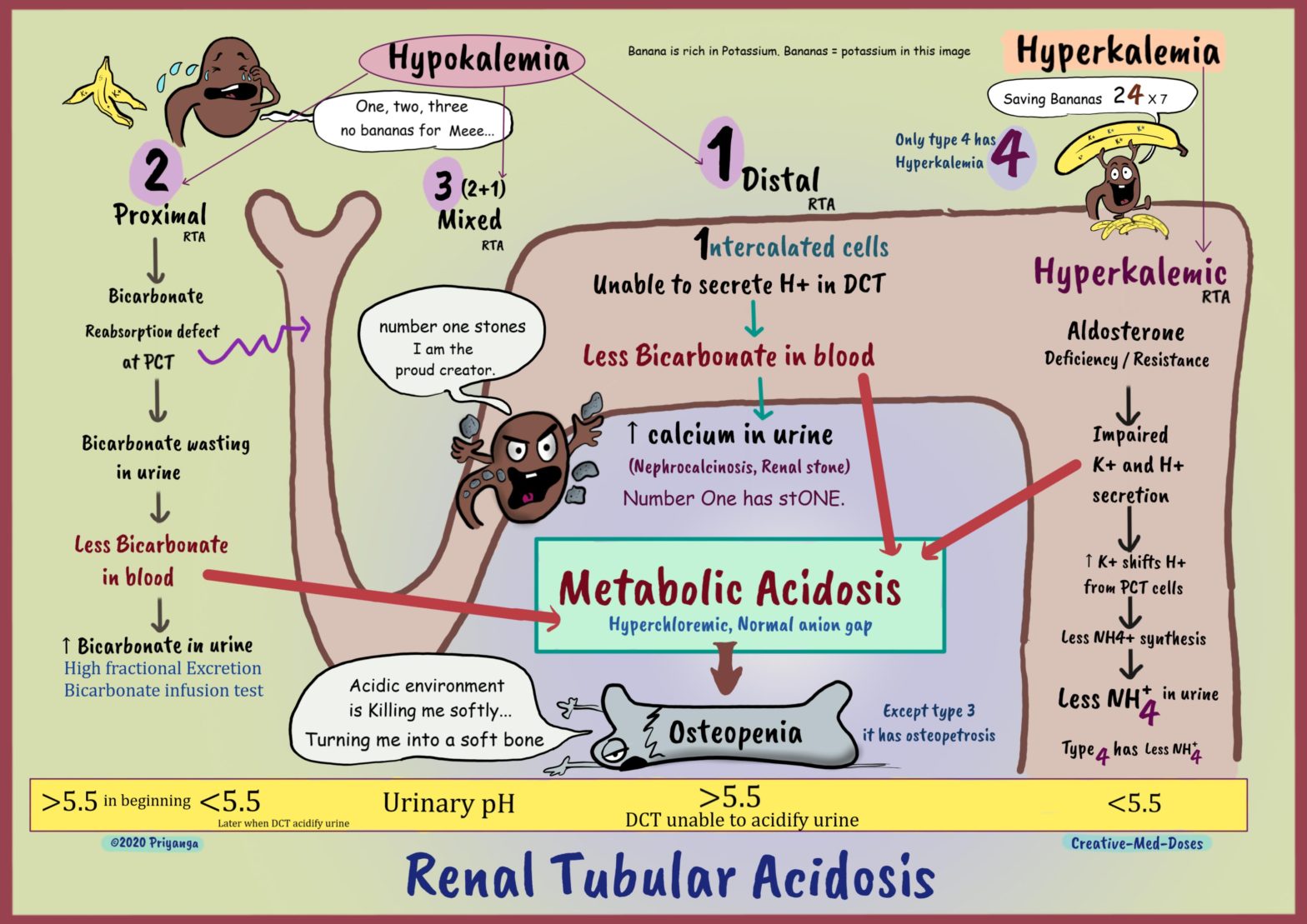
Renal tubular acidosis (RTA) is normal anion gap (hyperchloremic) metabolic acidosis in a patient with normal or almost normal renal function.
Patients with uremic acidosis (metabolic acidosis due to renal failure) have a decreased glomerular filtration rate (increased serum creatinine) and increased anion gap metabolic acidosis. Patients with renal tubular acidosis have relatively normal glomerular filtration rates and normal anion gap metabolic acidosis.
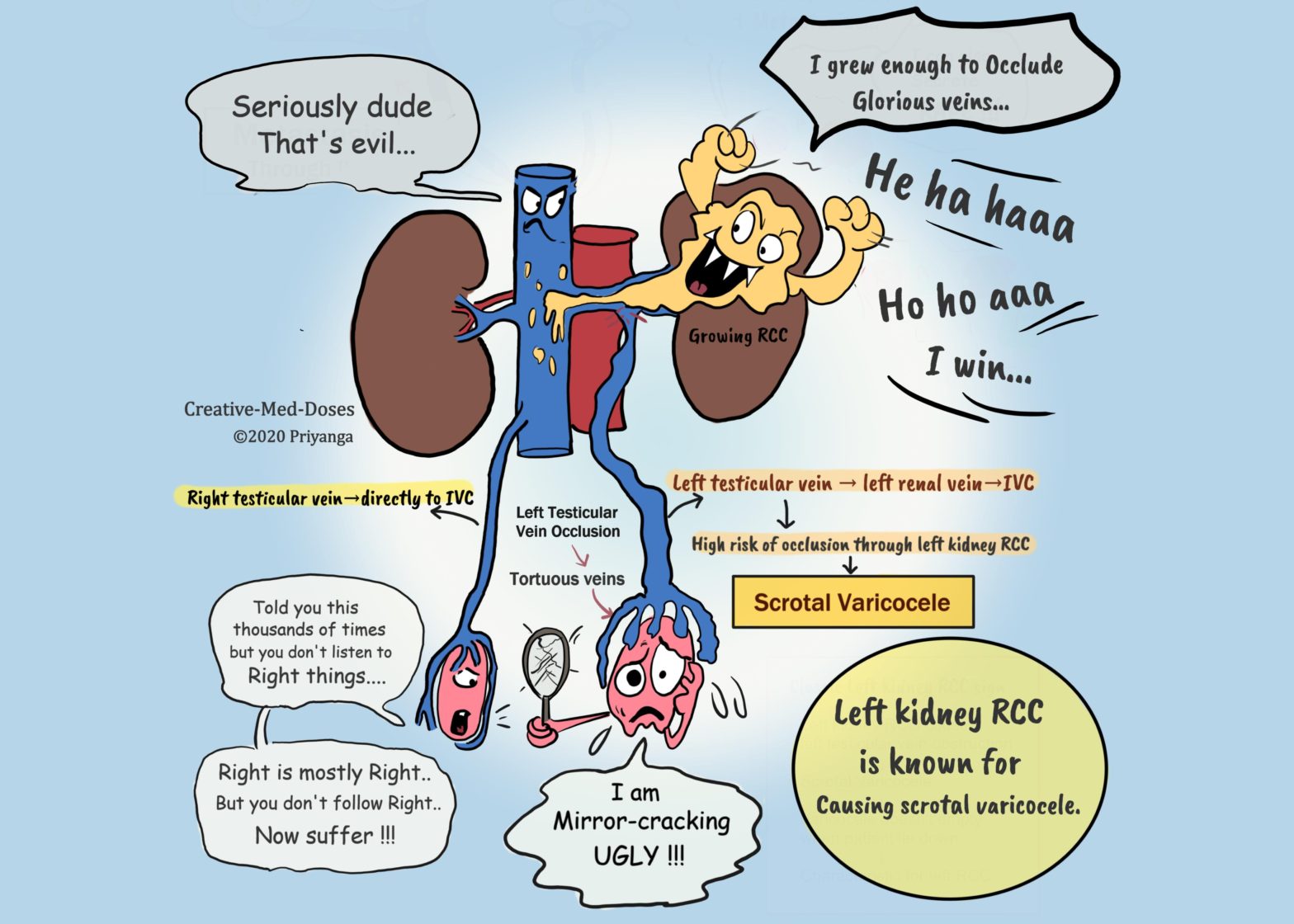
Renal cell carcinoma (RCC) arises from the renal tubular epithelium. It is the most common renal malignancy in adults (nearly 85% of renal cancers are RCC). RCC can be sporadic (in most cases) or associated with hereditary disorders.
Age on onset 6th -7th decade, more common in man. Hereditary renal cell carcinomas have an early age of onset.
High-risk group
Smokers
Hypertensive
Obese
Genetic factors
The acquired polycystic disease-The risk for developing renal cell cancer is increased 30-fold in individuals with the acquired polycystic disease.
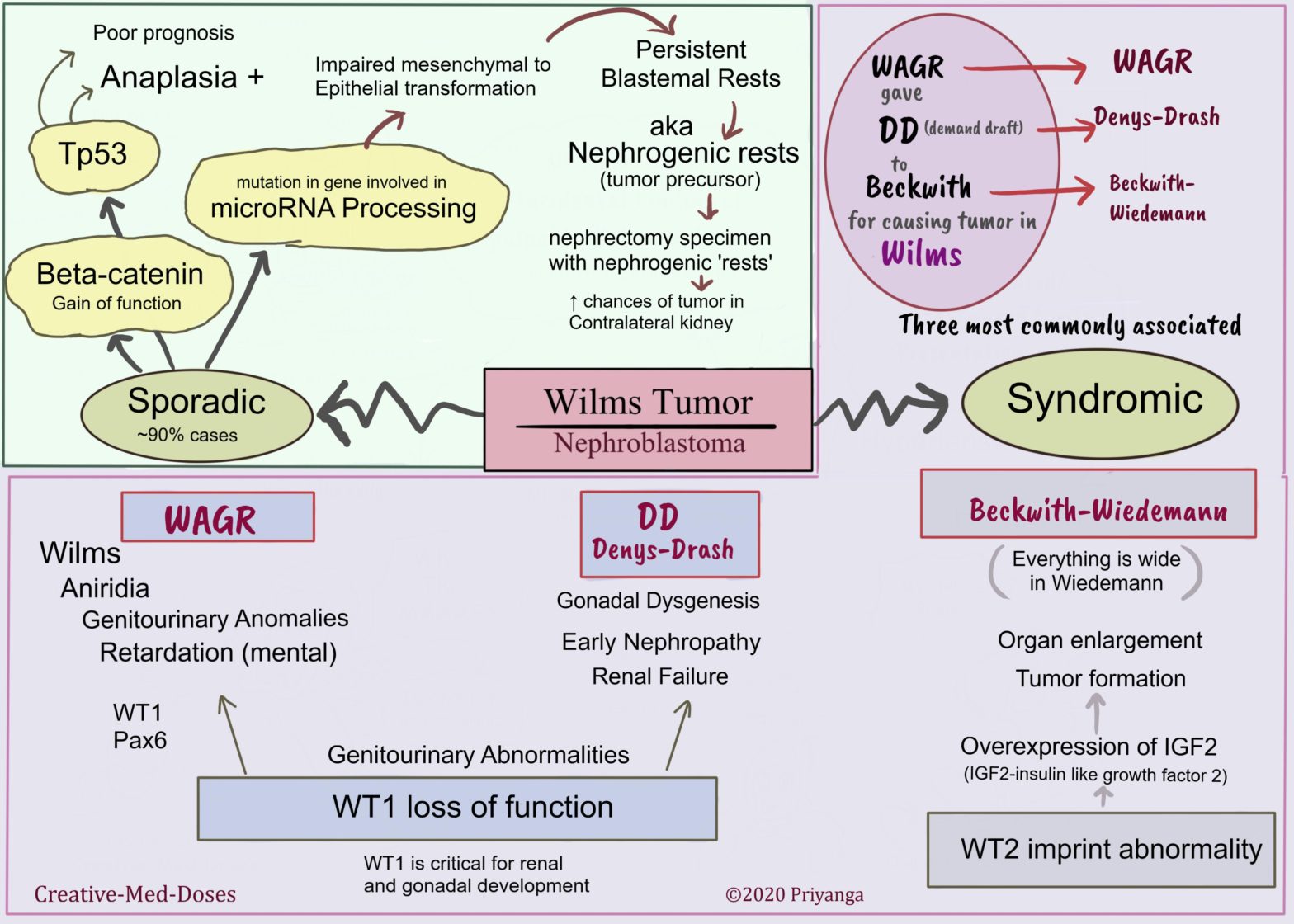
Wilms tumor (nephroblastoma) is the most common primary tumor of the kidney in children. Most cases are children between 2 and 5 years of age.
Pathogenesis
Sporadic (nearly 90%of cases) –
Molecular abnormalities related to sporadic cases are-
the beta-catenin gain of function mutation
Tp53 mutation
microRNA processing defect
Syndromic –
It is associated with other genetic mutations and hereditary conditions.
The following three are most commonly associated with Wilms tumor-
WAGR Syndrome-
Wilms tumor, Aniridia, Genitourinary abnormalities, and mental Retardation are present in this syndrome. It is associated with the deletion of the WT1 gene and Pax6 gene. The WT1 gene is critical for renal and gonadal development. The loss of WT1 causes renal tumors and genitourinary abnormalities.
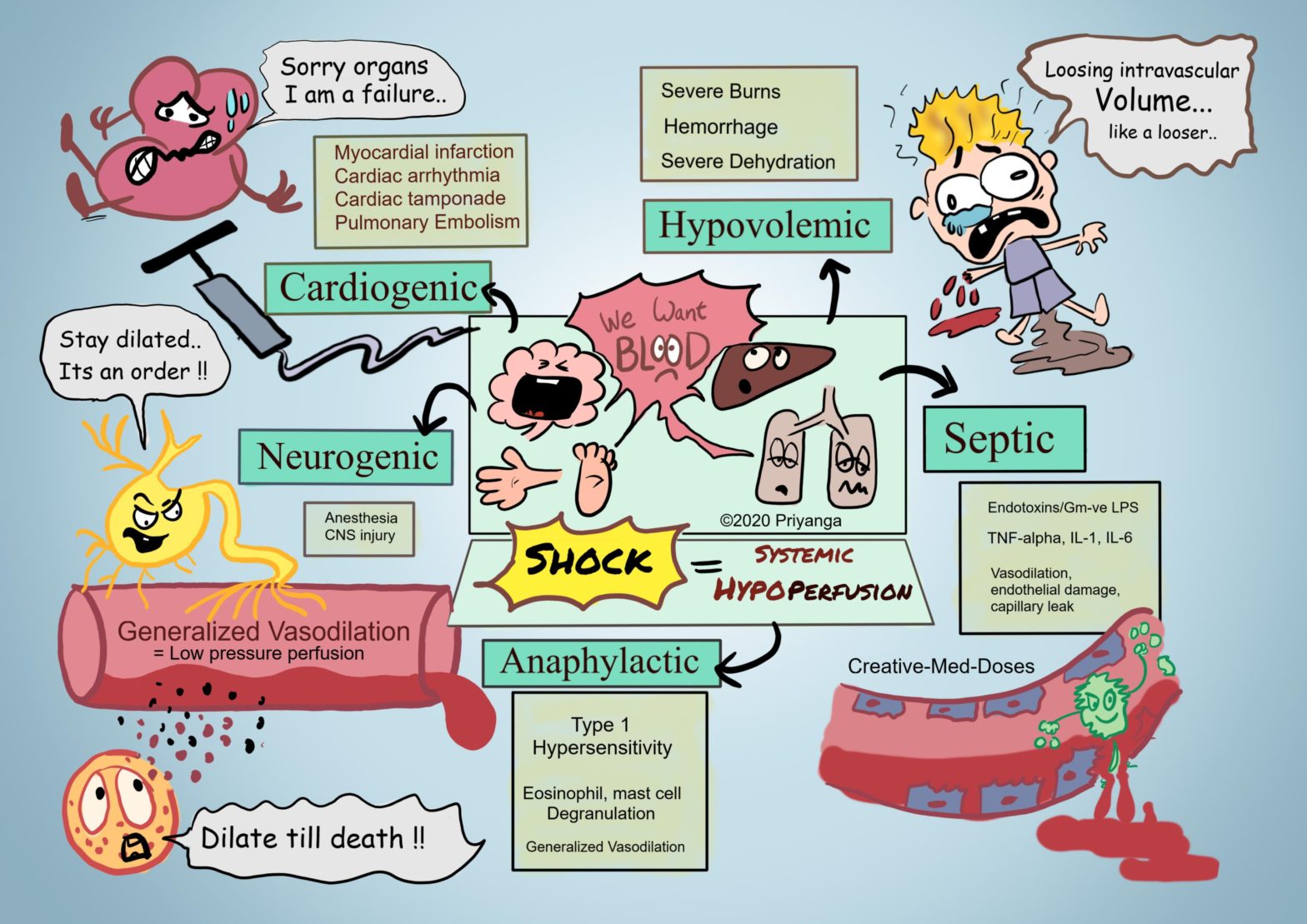
Shock is characterized by diminished cardiac output or reduced effective circulating blood volume which impairs tissue perfusion. This systemic hypoperfusion of various organs leads to cellular hypoxia and multiorgan failure.
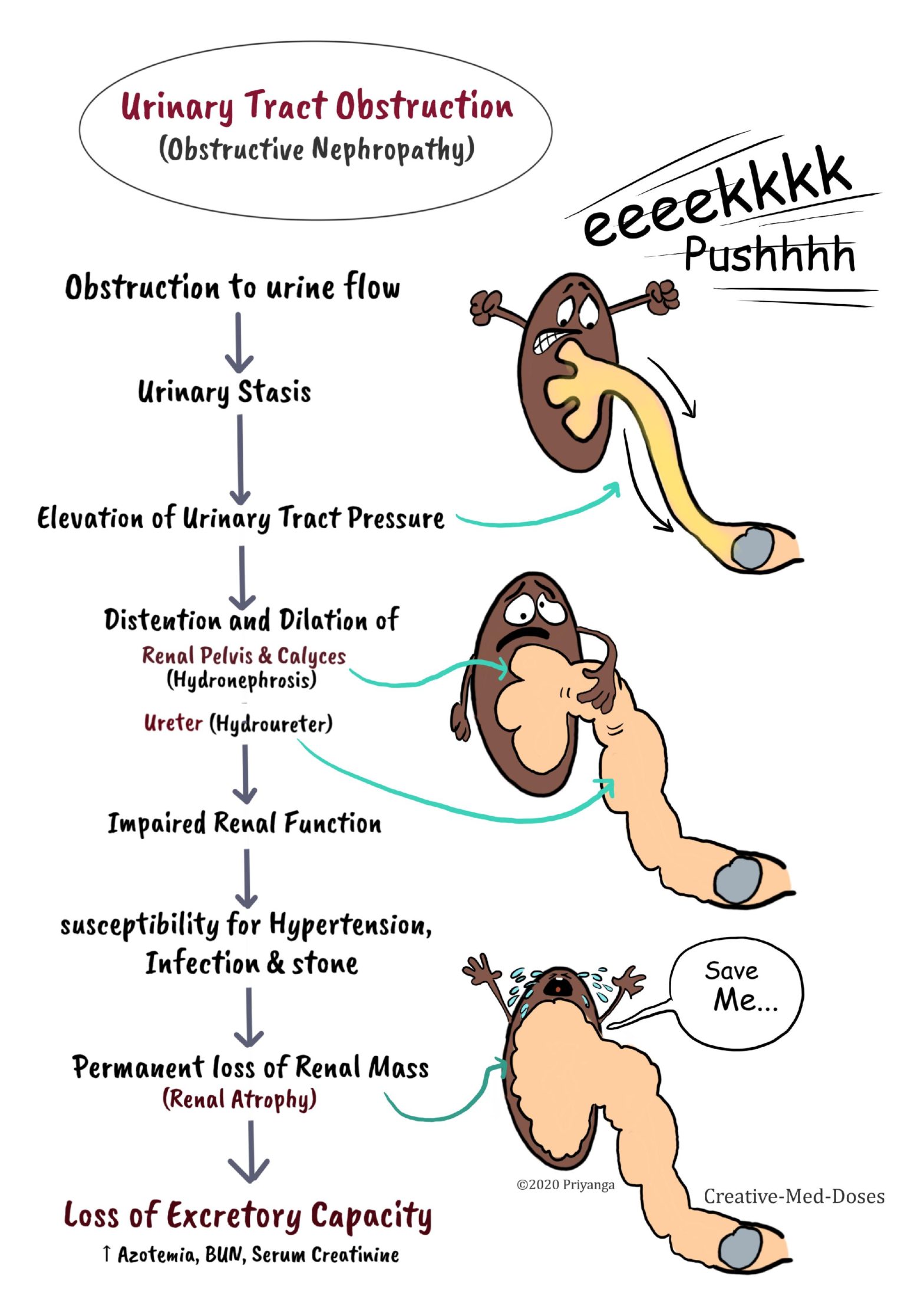
Urinary Tract Obstruction leads to impairment in the normal flow of urine → urinary stasis and increased pressure in part proximal to obstruction → increased pressure impairs renal and urinary tract functions → renal atrophy and cortical thinning. It is a common cause of acute and chronic kidney disease (obstructive nephropathy).
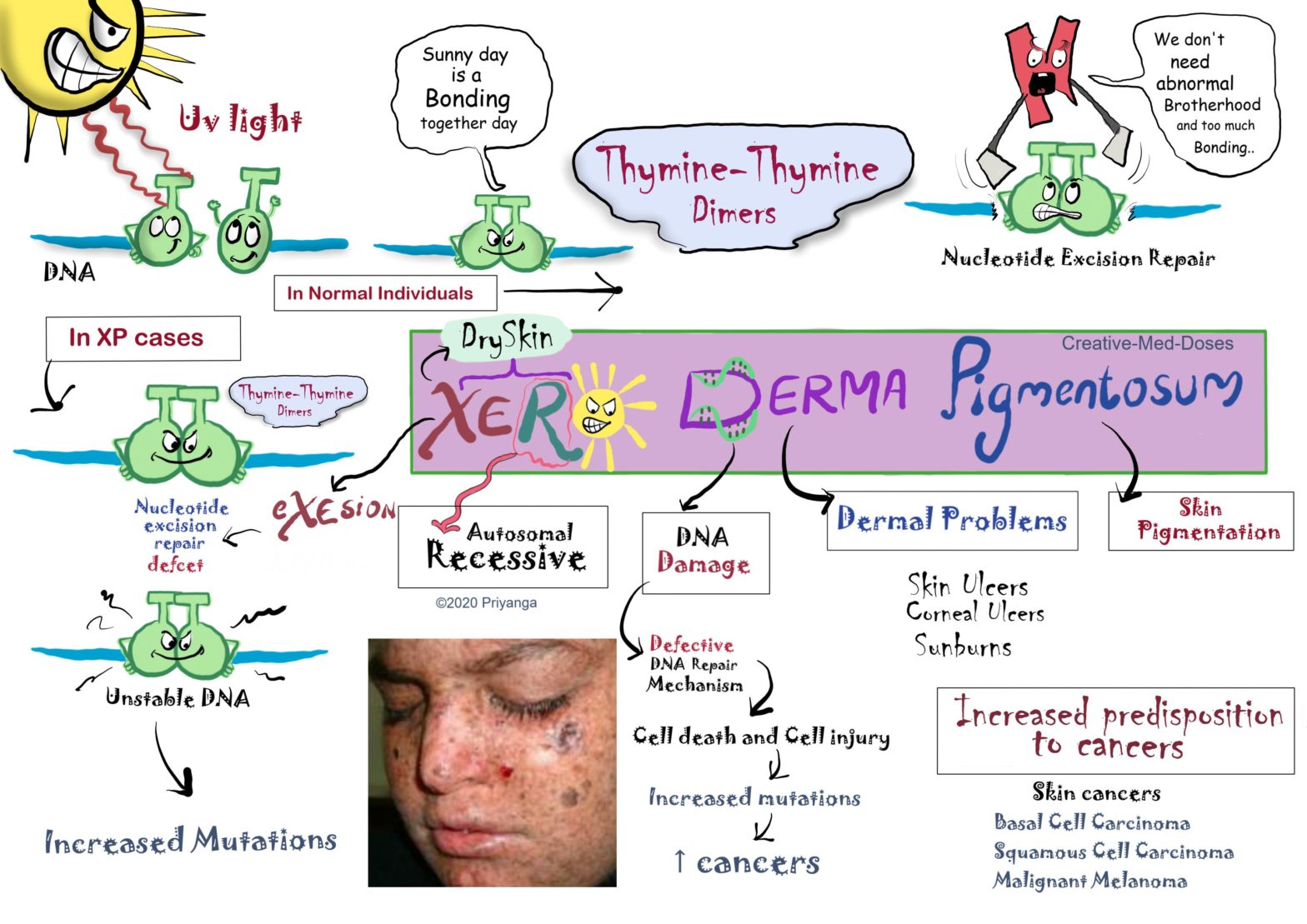
Xeroderma Pigmentosum (XP) is a rare, autosomal-recessive, hereditary skin disease. It is caused by defective DNA repair mechanisms (i.e., nucleotide excision repair). The affected individuals get skin ulcers and skin burns with minimal UV radiation (Sunlight contains UV Radiation so they can’t tolerate sunlight.