Budd-Chiari Syndrome (BCS) is a rare condition associated with obstruction of two or more major hepatic veins leading to hepatic venous outflow obstruction.
Etiology
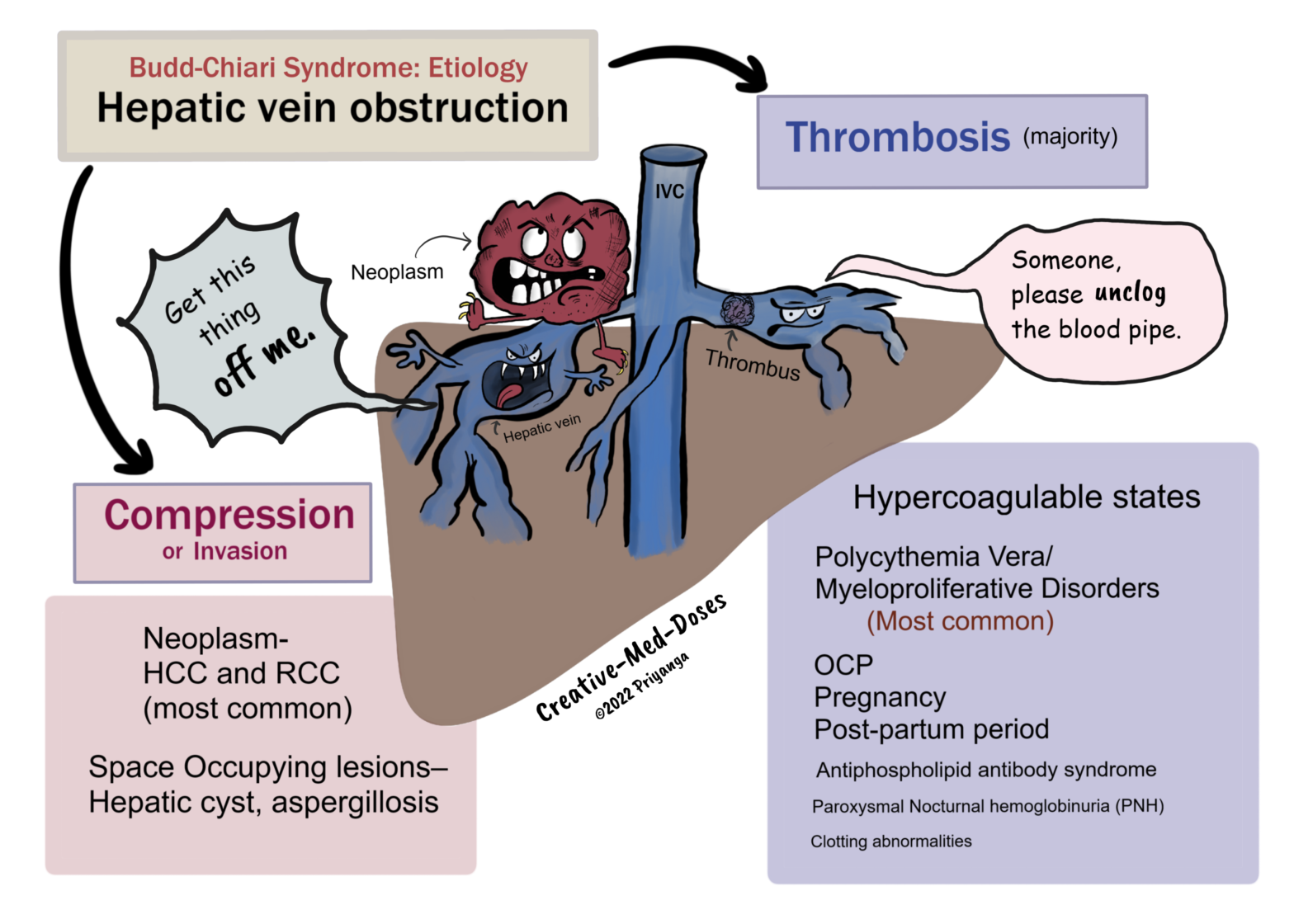
Hepatic Venous Outflow obstruction in Budd-Chiari Syndrome can be due to-
• Thrombosis of Hepatic veins
• Compression / Invasion of Hepatic veins
Thrombosis of Hepatic veins
The majority of cases with Budd-Chiari Syndrome are associated with hypercoagulable states causing thrombosis in hepatic veins. The most important causes are the following-
• Polycythemia Vera/ Myeloproliferative Disorders – nearly 50% of cases with Budd-Chiari Syndrome are associated with myeloproliferative disorders (Polycythemia vera is most common).
• OCP/ Pregnancy / Post-partum period – all are hypercoagulable states
• Antiphospholipid antibody syndrome
• Paroxysmal Nocturnal hemoglobinuria (PNH)
• Clotting abnormalities
Compression/Invasion of the Hepatic Vein
• Neoplasm- Intrabdominal malignancies (HCC and RCC are most common)
• Space Occupying lesions Liver – Hepatic cyst, aspergillosis
Pathophysiology
Obstruction of two or more hepatic veins →hepatic venous outflow obstruction→ impaired blood drainage to inferior vena cava→ increased backflow of the blood →hepatic venous congestion
Hepatic venous congestion→ hepatomegaly →stretching of liver capsule →abdominal pain and tender hepatomegaly
Hepatic venous congestion→ increases sinusoidal pressure → sinusoidal dilation →reduced hepatic blood flow→ cellular hypoxia→ centrilobular necrosis and peripheral fatty degeneration (Nutmeg liver) →liver dysfunction →jaundice, hyperbilirubinemia and increased liver enzyme→ liver failure if left untreated
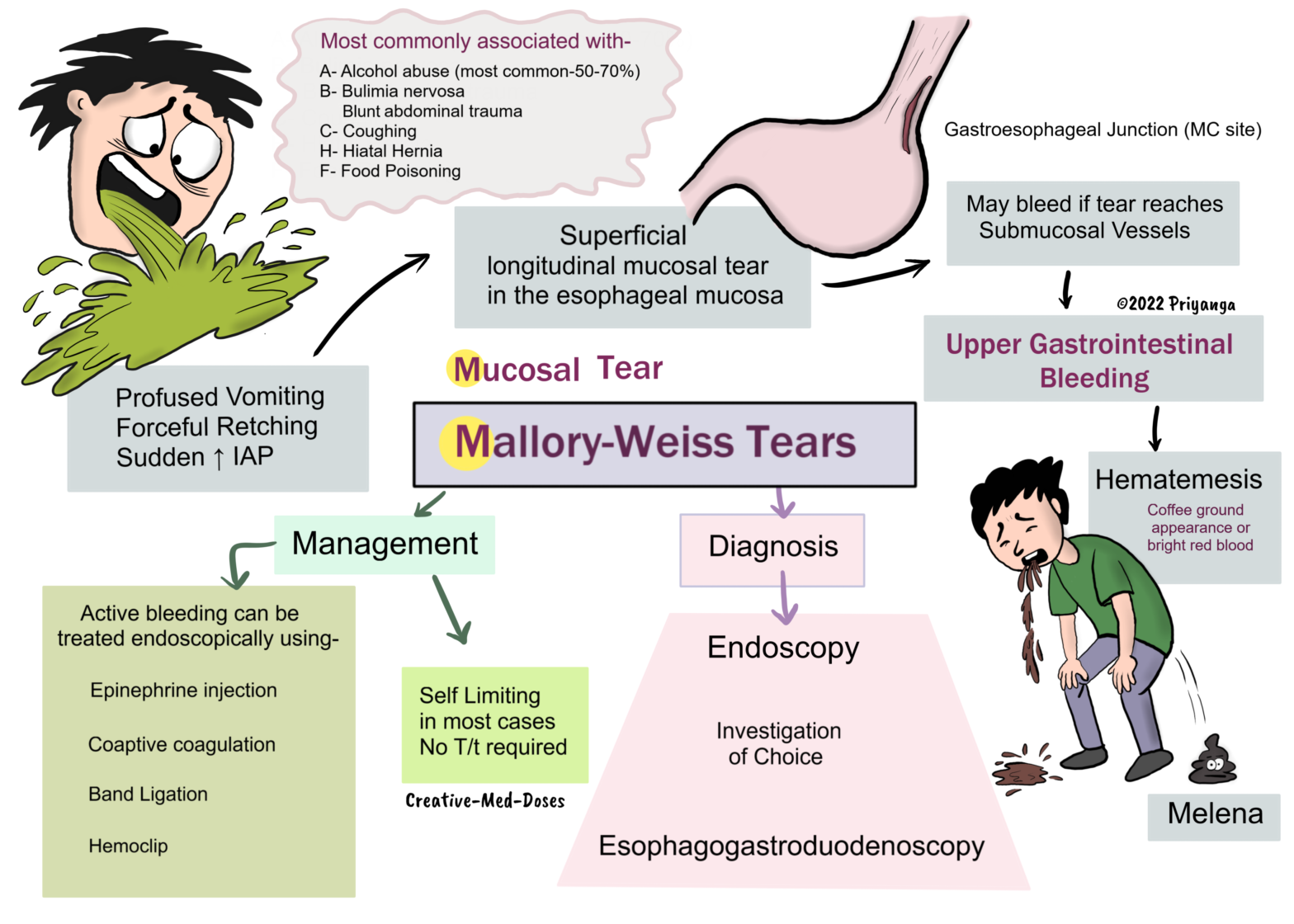
Mallory-Weiss Tears: Esophageal tear
Mallory-Weiss tears are longitudinal mucosal tears in the esophageal mucosa. It is one of the common causes of acute upper gastrointestinal bleeding, and the gastroesophageal junction (gastric cardia) is the most common site.
The tear is superficial and involves only the mucosal layer, in contrast to Boerhaave’s syndrome. Boerhaave’s syndrome has full-thickness rupture (the tear involves the muscular layer).
These tears most commonly occur after the period of profuse vomiting, violent coughing, or retching and result in a short period of hematemesis.
The tear is caused by repeated acts of a sudden increase in intraabdominal pressure (IAP). The increased intraabdominal pressure is associated with retching, vomiting, straining, coughing, cardiopulmonary resuscitation (CPR), and blunt abdominal trauma.
Most commonly associated with
• Alcohol abuse (Most common 50-70%)
• Eating disorders associated with forceful retching (bulimia nervosa)
• Food poisoning
• Hiatal hernia
Pathogenesis
The sudden and rapid increase in intraabdominal pressure → Reflux of acid with pressure → Fluid flow shear stress → Longitudinal tear in mucosa → Tear may reach deep into the submucosal arteries and veins → Upper GI bleeding (Hematemesis).
The rapid increase in intraabdominal pressure may happen in-
• forceful and repeated emesis (Bulimia)
• profuse vomiting (alcoholic abuse)
• coughing
• blunt abdominal trauma
Clinical presentation
• Hematemesis- coffee ground appearance or bright red blood
• Epigastric pain
• Melena
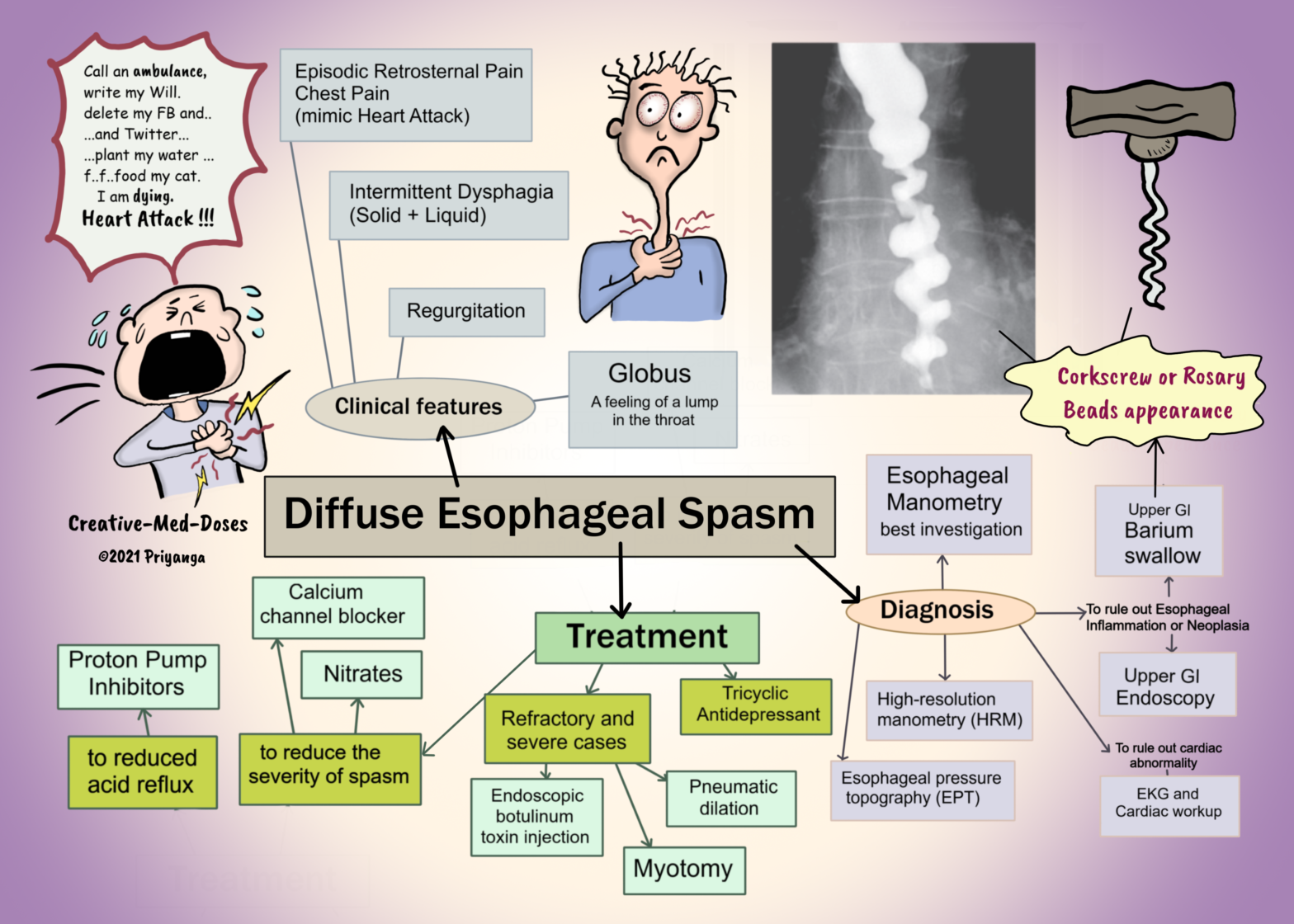
Diffuse Esophageal Spasm (DES) is a rare motility disorder of the esophagus. It is characterized by diffuse non-peristaltic, uncoordinated spasmodic contractions in the esophagus. It leads to episodic retrosternal chest pain and intermittent dysphagia.
Etiology
There is an imbalance in the coordination of the inhibitory and excitatory postganglionic pathways. It leads to non-peristaltic spasmodic contractions. Several segments of the esophagus contract independent of each other simultaneously, causing improper propagation of the food bolus in DES. It causes intermittent dysphagia, retrosternal chest pain, regurgitation, and globus sensation.
The exact cause is unknown. The following are likely to be associated with DES-
• Acid reflux in gastroesophageal reflux disease (GERD) irritates the esophageal wall and may trigger a diffuse esophageal spasm
• Neurological disorders (neurodegenerative/ neuromuscular disorders) may lead to an imbalance between inhibitory and excitatory signals. It causes uncoordinated spasmodic contractions.
• Increased release of acetylcholine
• nitric oxide-mediated impairment of inhibitory ganglion neuronal function
• high BMI and high cholesterol levels might cause lower esophageal sphincter dysfunction and GERD
• Anxiety or depression
Clinical presentation
Episodic retrosternal chest pain can mimic heart attack/ angina. It might manifest upon eating quickly or consuming very hot or cold food.
Intermittent dysphagia- can be associated with both solid and liquid food.
Globus sensation – feeling a lump in your throat as if something is stuck in the throat.
Corkscrew or rosary beads appearance on barium study (only during episodic spasm and pain).
Kidney does not have a very energy-efficient waste disposal system.
First, the glomerulus filters all the sodium, nutrients, and water needed for the body and sends them to the tubular lumen.
Then, the tubular epithelium reabsorbs most of these nutrients (including sodium) and water. The reabsorption of sodium is an active process. It consumes ATPs as an energy source. The oxygen requirement of the kidney is high because of the ATP consumed in active reabsorption.
The oxygen demand of the proximal tubular segment and thick ascending loop of Henle is very high. This high demand makes these parts of the renal tubule vulnerable to hypoxia and acute kidney injury.
What kind of waste disposal system first throws waste along with precious nutrients and then spends loads of energy and time to bring those nutrients back in the blood. Sounds stupid and inefficient!
Our kidneys are not as energy-efficient as we think, but yeah, they are crazy busy. We can at least try not to burden them more by avoiding the consumption of excess salt and sugar.
Renal hypoxia can lead to acute kidney injury and chronic kidney disease.
The kidneys weigh less than 0.5% of body weight and receive 20%–25% of our cardiac output at rest. The kidneys are the most highly perfused organs in the body if we calculate blood per gram of the tissue.
Despite having an ample supply of blood, the kidney especially, the renal medulla is highly vulnerable to hypoxia and subsequent ischemia. Hypoxia and ischemia are the significant pathophysiological features of acute kidney injury and chronic kidney disease.
The S3 segment (straight segment) of the proximal tubule and the medullary thick ascending limb of the loop of Henle (mTAL) are most prone to ischemic injury. Both these tubular areas exist in relatively lower oxygen conditions. It is a trade-off to be able to get a high osmotic gradient and concentrated urine.
1.Renal tubules have high metabolic demands due to active reabsorption of sodium -Metabolic demands of renal tubules keep them hungry for oxygen, especially proximal convoluted tubules of the renal medulla are always on the verge of getting hypoxia.
The proximal convoluted tubules reabsorb most of sodium and water from tubular filtrate. Sodium reabsorption is an active process that requires energy in the form of ATP. The production of ATP is dependent upon oxygen supply. PCT cells are always in demand of oxygen, and anything which compromises the oxygen delivery to these cells causes PCT cell injury and death.
Capillary Leak Syndrome is associated with an increased capillary permeability to proteins. It leads to the loss of protein-rich fluid from the intravascular space to the interstitial space.
Capillary leak syndrome is characterized by
• diffuse pitting edema,
• exudative serous cavity effusions,
• noncardiogenic pulmonary edema,
• hypotension
• hypovolemic shock with multiple-organ failure (in severe cases)
Following conditions can lead to increased capillary permeability and CLS-
• Sepsis (most associated) Idiopathic systemic capillary leak syndrome (SCLS) or Clarkson’s disease
• Engraftment syndrome
• Differentiation syndrome
• Ovarian hyperstimulation syndrome (OHSS)
• Hemophagocytic lymphohistiocytosis (HLH)
• Viral hemorrhagic fevers (VHFs)
• Autoimmune diseases
• Snakebite envenomation
• Ricin poisoning
• Drugs (monoclonal antibodies e.g. rituximab)
Pathophysiology
Most diseases causing capillary leak syndrome have a similar underlying pathophysiologic abnormality—an increased capillary permeability to proteins.
Hypercytokinemia → Adherens junction and tight junction disruption → capillary endothelial damage and disruption → capillary become permeable for proteins→ loss of protein-rich fluid→ reduced intravascular volume and increased interstitial fluid → sign and symptoms of capillary leak syndrome.
Clinical Presentation
Hemodynamic manifestations
Capillary leak → Loss of protein-rich fluid from the intravascular space→ intravascular volume depletion → secondary activation of the renin, angiotensin, and aldosterone system →sodium and water retention → systemic edema and exudative serous cavity effusions and ascites.
Loss of intravascular volume can cause hemoconcentration in severe cases. And abrupt and significant fluid loss through the intravascular compartment. The hemoconcentration can be used as an indicator of capillary leak severity.
The capillary leak syndrome can lead to hypovolemic shock and acute kidney injury in severe cases.
Carbon monoxide (CO) is an odorless, tasteless, and colorless gas that can cause sudden illness and death if inhaled.
CO is generated in the incomplete combustion of carbon compounds. And the common sources include fire, engine exhaust, and faulty furnaces.
Risk factors
• Wood burning heaters
• Poorly ventilated buildings
• Use of charcoal, gas, or petroleum
• Faulty furnaces
• Motor vehicle exhaust
• Inhalation of methylene chloride from paint thinners
Pathogenesis
Absorption of inhaled carbon monoxide occurs in the gas exchange region (alveoli) of the respiratory tract following inhalation.
It displaces oxygen from hemoglobin and causes tissue hypoxia.
Most carbon monoxide binds reversibly to hemoglobin (Hb) in red blood cells and other heme proteins. Carbon monoxide’s affinity for hemoglobin is 200–250 times greater than that of oxygen.
After binding to Hb to displace oxygen and form carboxyhemoglobin, carbon monoxide is transferred rapidly throughout the body, where it causes cellular hypoxia and asphyxia.
Dissociation and excretion of carbon monoxide occur rapidly after cessation of exposure.
Binds to cytochrome oxidase and impairs electron transport chain leading to cellular hypoxia and reduced ATP generation, cell injury, and cell death.
Cardiovascular injury can result from carboxymyoglobin formation and vasodilation from the cellular effects of carbon monoxide.
Clinical neurological effects and any delayed neurological complications are due to cellular hypoxia. The cellular lipid peroxidation also increases neuronal injury, brain edema, and neurological dysfunction.
How much is too much?
According to the World Health Organization, levels greater than six ppm are potentially toxic over a longer period of time.
The COHb levels of 2% or more in nonsmokers and 10% or greater in smokers are considered potentially harmful and likely to produce symptoms.
Clinical Signs and Symptoms
Headache
Dizziness and confusion
Nausea and vomiting
Shortness of breath
Altered mental status
Cherry-red skin (late finding)
Coma, Loss of consciousness, Death
Cardiovascular complications
• Myocardial injury
• Myocarditis
• Arrhythmia
Neurological complications
• Impaired memory
• cognitive dysfunction
• depression
• anxiety
• vestibular and motor deficits
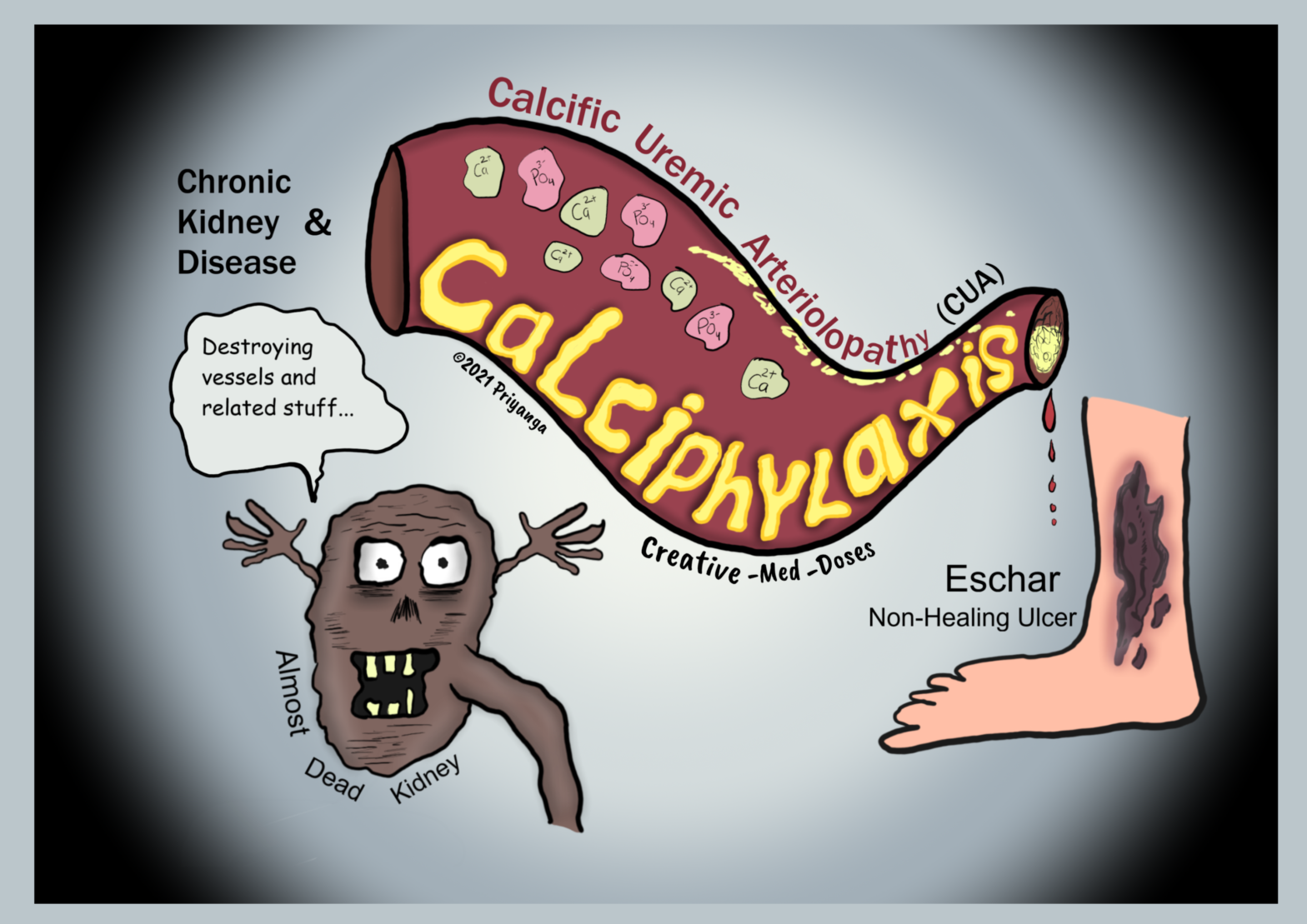
Calciphylaxis or calcific uremic arteriolopathy (CUA) in chronic kidney disease is a rare but fatal complication seen in patients in the late stage of chronic kidney disease.
The calcification of small vessels, especially arterioles, is a characteristic feature. And it eventually leads to the obstruction of the blood flow.
The blood flow obstruction causes ischemia, necrosis, severe pain, and color changes at the site of obstruction.
Pathophysiology
Calcific uremic arteriolopathy (CUA) has a multifactorial pathogenesis.
• The imbalance between inducers and inhibitors of calcification
• Increased use of oral calcium as a phosphate binder
• Secondary hyperparathyroidism
• Hyperphosphatemia – elevated serum phosphate induces a change in gene expression and switches vascular cells into osteoblast-like cells. These cells cause vascular calcification.
• Uremia in end-stage renal failure causes inflammation and suppression of calcification inhibitors. Suppression of calcification inhibitors leads to more calcification and obstruction.
• Warfarin in dialysis patients – Warfarin used in hemodialysis decreases the vitamin K–dependent regeneration of matrix GLA protein. The matrix GLA protein is crucial in preventing vascular calcification. That’s why warfarin treatment is considered a risk factor for calciphylaxis. If the patient develops calciphylaxis, discontinue warfarin and replace it with another anticoagulant.
The sequence of events in calciphylaxis
vascular calcification→ luminal narrowing→ ischemia →skin necrosis and ulceration. If left untreated, it can cause secondary bacterial infection. Which can lead to sepsis, septic shock, and death.
Clinical presentation
In the early stage of the disease, the patient presents with pruritus and cutaneous laminar erythema or a violaceous rash. It may resemble livedo reticularis.
In the late stage of disease, cases have painful eschars and painful non-healing ulceration and necrosis.
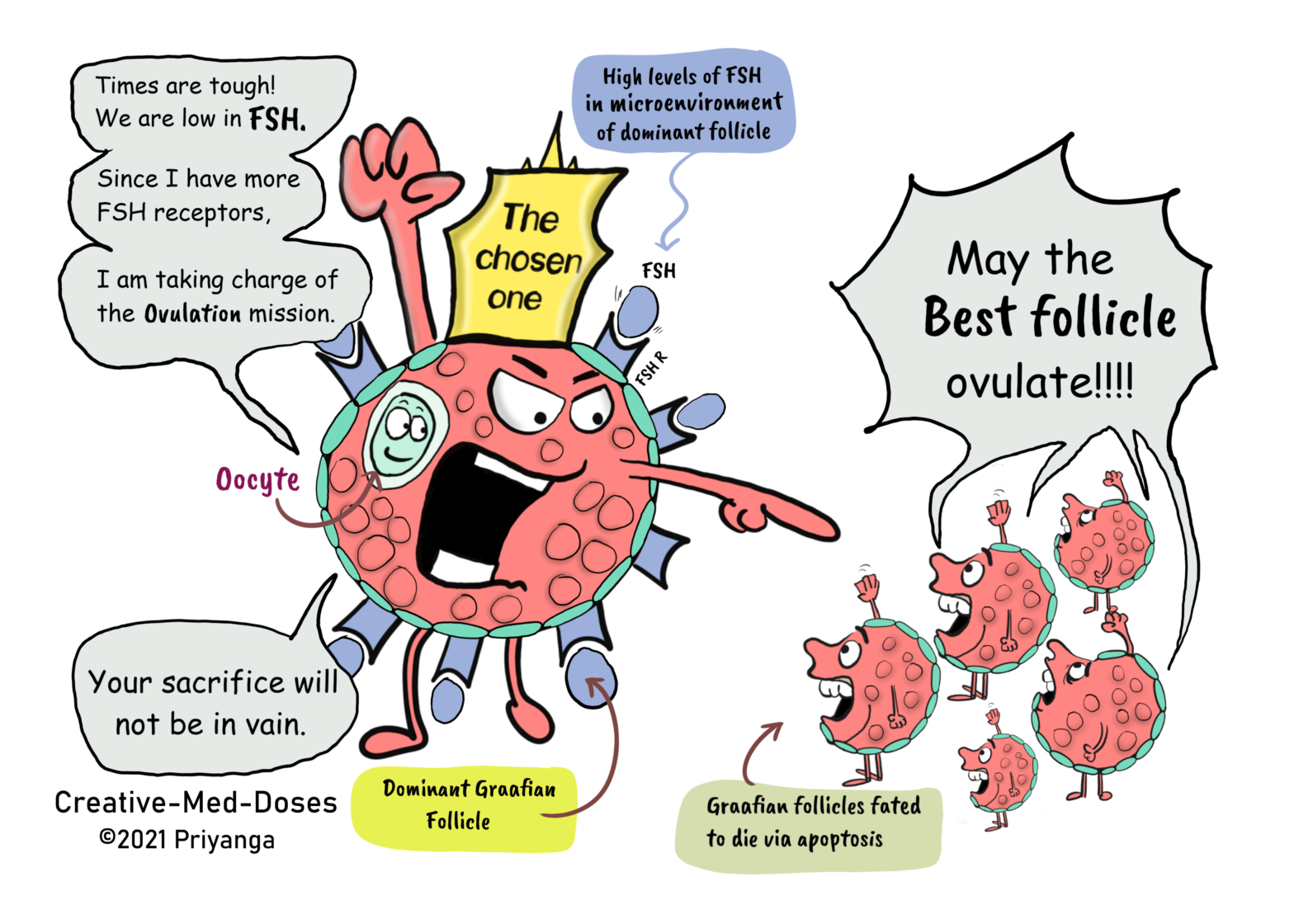
In every menstrual cycle, the ovaries select and induce the growth of a single dominant follicle that participates in single ovulation. The selection of the dominant follicle is under the control of the hormones FSH and LH.
Any interference directly or indirectly with the normal action of the gonadotropins can lead to apoptosis of follicles and may cause infertility.
Morphometric analysis of healthy ovaries showed that the dominant follicle which ovulates in the subsequent cycle is selected from healthy follicles measuring 4.7 ± 0.7 mm in diameter at the end of the luteal phase of the menstrual cycle. The selection of the dominant follicle occurs at the late luteal phase of the menstrual cycle.
The dominant follicle has a high rate of mitosis in the granulosa cells, it is the characteristic feature of the dominant follicle.
When one follicle is selected, the granulosa cells in the chosen follicle continue dividing at a relatively fast rate. And proliferation slows in the granulosa cells of the other follicles. These follicles will become atretic follicles eventually (dies via apoptosis).
How the dominant follicle is selected
The secondary rise in plasma FSH is a must to find/select dominant follicles.
The secondary FSH rise in women begins a few days before the progesterone levels fall to basal levels at the end of the luteal phase. And the FSH levels remain elevated during the first week of the follicular phase of the cycle.